谈谈学量子化学如何正确地入门
PS: 如果你是从零起步的初学者,想入门量子化学,那么恭喜你,看到此文章时,你终于找到正轨了!
How to get started correctly in quantum chemistry
文/Sobereva @北京科音
First release: 2016-Nov-12 Last update: 2025-Dec-27
笔者认为量化入门一定要按照正确、合理的顺序,循序渐进。要从简单到复杂,从构建整体的知识框架并会用最常用的量化程序算最基本的任务开始,再到逐渐了解更多理论、深化对理论的理解以及掌握更多计算技巧和程序的使用。在这个过程中,参加合适的培训、看合适的资料尤为关键。参加不适合自己当前程度的培训、看不适合自己当前水平的书,不仅没什么益处,花很多时间也学不到自己当前需要的知识,反倒会有严重的害处,就是会接触到各种复杂的概念、术语、公式,自己事先又没有足够能够理解它们的基础知识,就会被这些东西搞晕,甚至胡思乱想,把五花八门不相干的概念瞎搅合在一起。
有两类初学者,他们是极度危险的:
第一类初学者:决心要把量化的理论学明白,还打算把涉及到的各种公式推导全都搞透,想等彻底学明白了才肯研究实际问题。甚至有些人,在学量化理论前,还觉先得把数学、物理再好好学一遍,否则觉得心里没底。这类初学者,除非迷途知返,否则后果是,轻则把导师惹怒(课题迟迟没进展,都不知道人整天在干什么),重则延期毕业。因为这种方式入门的周期实在太漫长,而且空学一堆理论不实际上阵算算具体问题,就只会纸上谈兵、不知哪些知识重要哪些不重要、哪些有用哪些没用,而且也没法通过实践加深对理论的理解。而且费劲巴拉把一大堆数学推导什么的好不容易都弄明白了,在之后自己的研究中它们又完全派不上任何直接的用场(除非是专门搞基础性理论方法研究的),后果就是很快会把它们忘记,搞懂它们花的精力几乎全都白费了。这些初学者一定要意识到学习的时间成本问题,时间不是无限的,毕业、文章等各种现实压力在眼前,没有时间让你无限制地悠哉悠哉地慢慢啃理论。量化历史悠久,博大精深,深坑无数,很多理论方法对于应用性计算来说,实际上只要正确地知道个大概便足够,如果无论用到什么理论都非要深究一番,那真是在有限的时间内什么也干不成了。比如十分流行的M06-2X泛函,知道它的一些关键性特点(Truhlar组08年搞的、是meta-杂化泛函、对有机体系进行参数化、对积分格点略敏感、算热化学数据/弱相互作用等问题优秀...),然后放心用它就够了,若是非要把原文看明白,然后发现看不明白又去看DFT专著,然后为了加深对泛函的理解又买本《泛函分析》啃,那得猴年马月才能开展研究。
第二类初学者:这类初学者严重忽视理论基础的重要性,什么都不懂上来就看网上零七八碎的信息来用量化程序做计算。前面说了在初学阶段花过度时间用在啃理论是不对的,但是量化上的常识知识不能不知道!没有常识就能算出合理、有价值、能用来发表的数据才怪!很多初学者,严重缺乏基本常识,犯各种低级错误,令高手感到莫名其妙、匪夷所思的各种小白问题基本都是这类初学者问的。这些初学者的思路、对被研究的问题的看法往往都还处在民科阶段,写出来的关键词令人无语,诸如用热力学组合方法的时候还写基组名。而且网上(除了计算化学公社和思想家公社群外)净是乌七八糟误人子弟的量化方面的讨论,还有各种错误百出的资料(大多数是中文的)。这些初学者看了这些内容,会学一身坏毛病、获得严重错误的知识经验,乃至陷入泥沼不能自拔,诸如到处用IOp(5/13=1)、靠加大循环次数上限妄图解决不收敛、明明程序支持解析频率非要加个=numer用数值的。总之这类初学者的下场就是,会计算、能算出来数据,但绝大多数的计算都是白费功夫、糟蹋计算资源,高手能用不足他们的1/10甚至1/100的计算量算出合理、有意义得多的结果。
综上所述,在量化入门过程中,理论知识和运用实际程序计算,二者是相辅相成的,切不可彼此脱离。没有常识级别的理论知识,算就是瞎算;不用量化程序计算具体问题,一方面完成不了科研任务,另一方面也无法深化对理论方法的认识。
那么,如何以最正确的入门途径踏入量化研究领域?分两种情况,一是有条件能参加北京科音量化培训的情况,二是没条件只能自学的情况:
一 参加培训
参加笔者讲授的北京科音自然科学研究中心(www.keinsci.com)开办的不同档次的量子化学培训,参加此培训是入门和提高的非常好而且特别快速的途径。靠自学学习不仅入门慢得多得多,更糟糕的是看五花八门的资料过程中往往还会被错误的信息误导,并且很难形成正确的知识框架。
北京科音的量子化学培训从2015年起开办,共分为三个班:初级班、中级班、高级班,内容难度、深度依次增加。每年三个班都会在北京开办,培训的预告在北京科音的首页上可以看到,每次培训前大概一个月左右会在主页上发正式通知。历届培训的记录可以在北京科音官网的“科研培训”中看到,在计算化学公社论坛还可以看到学员们参加历届培训的感想:http://bbs.keinsci.com/forum-43-1.html。培训开办目的是让广大量化工作者最大程度受益,所以培训费用仅有其它机构平均培训费用的不到一半,而对培训质量的要求则是最高级别、最苛刻的,这使得每次培训都赢得了学员们热烈的反响,场场爆满。有很多学员甚至从澳大利亚、美国、德国、沙特阿拉伯、韩国、加拿大、以色列等国专程回国慕名参加。北京科音举办的量子化学培训班已经成为了大量中国年轻一代量子化学研究者的摇篮。学员从零基础开始,参加初级班后可以成为中鸟,经过一段时间研究实践后,再参加中级班后可以成为老鸟(对于悟性好的,把中级班所讲知识完全吃透就达到高手程度了)。如果对量化颇有兴趣,感觉仍学有余力,或者想获得研究更深层、更多问题的能力,那么再参加高级班,就可以达到大侠的程度。
这些培训的下一届举办时间见北京科音官网首页的预告栏。等不及下一次培训者,不方便到场参加者,或者报名没抢上者,也可以购买往届培训纸质讲义+全程录音+电子资料自学,详见下面给出的相应培训班的介绍页面链接。
下面分别介绍北京科音的初级、中级和高级量子化学班的特点和定位:
初级量化班:
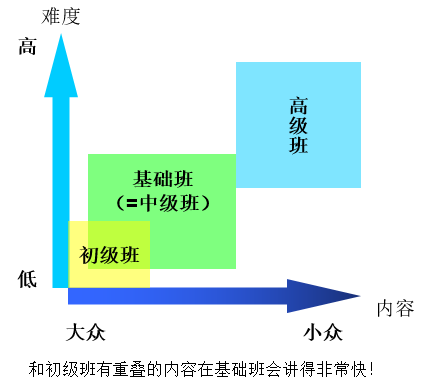
初级班时长为4天,从2016年12月开始办第一届,平均每年办两届。这个班专门针对广大从零基础开始入门的量化研究者,还有那些接触过一点量化计算,但头脑里稀里糊涂,各种常见任务都不能确保正确完成的人。这个班也适合那些实验化学工作者,他们需要学习量化计算来辅助自己的实验,但不打算也没时间精力把量化计算学得很精通。参加过此培训后,学员就可以不犯错地顺利完成各种常见量化计算任务,如几何优化、找过渡态、计算热力学量、势能面扫描、算NMR/红外/拉曼/UV-Vis谱等等,而且也会学会所有最最基础的量化理论知识,确保有足够的常识。初级班是精心设计的给初学者入门的最佳捷径,对于那些刚开始搞量化的,身边又没真正懂行的人教,上过初级量化班后一定会感觉这个班真是太难得了。也确实,参加此培训后的学员们整体反响极好。初级班的ppt在800页左右,初不仅把问题掰开了揉碎了慢慢讲,还留出时间辅导学员上机操作,以确保每一个学员都能掌握全部所讲内容、能亲自算相应问题,基本不会有听不懂的地方。如果上初级班后,又自己做了一阵子量化计算,感觉初级班知识都掌握得很好了,对量化计算又有兴趣,想学得更多、掌握更多知识技巧,使自己能运用量化方法更好、更快地研究更多问题,那么绝对不要错过中级班。为了系统性和完整性,中级班有1/4左右的内容和初级班有重叠,但重叠的部分会讲得很快。
初级量化班讲授的详细内容介绍见:http://www.keinsci.com/workshop/KEQC_content.html。
值得一提的是,有些学校开设了量子化学课,但如果量化课是那种讲一堆老掉牙理论的那种,哪怕你考试得了满分,量化计算方面实际上也还属于本质上零基础状态。如果是那种有点实例的量化课,由于学校里开的那种课讲得一般都非常浅,涉及的东西很少,内容不系统,所以只是参加过那种课的程度,参加初级班也是极度重要和非常有帮助的。
中级量化班:
这个班内容超超超级丰富,时间为6天现场+29个小时的视频(折合6+3.5天),总共70多个小时,ppt多达超过2600页!培训把所有常见量化问题的原理和计算方法全都照顾到了,是其它地方很难学到的。许多主题,比如(超)极化率计算、弱相互作用计算、分子力学与ONIOM、周期性计算、反应速率计算、MOPAC的使用等等都是初级班里没有的,而且对于初级班已有的主题,比如量化的理论基础、基组、几何优化、找过渡态、热力学量的计算等等,会讲得明显更深入、更全面,传授更多高级的经验技巧。中级量化班由浅入深讲得很系统,也很明白,只要课前预习,课上仔细听,再加上回去反复复习讲义N遍,能力不错的学员是能完全消化的,并可以达到理论功底扎实、计算操作上炉火纯青的程度。但是中级班有一定难度,而且信息量巨大,根据之前的培训经验,零基础的学员往往跟不上,接受能力差的也就能吸收、领会所讲内容的1/3甚至更少。另外,中级班信息量大而时间相对有限,所以讲得较快,且虽然提供了大量实例和练习,但没时间对那些很初级的操作做过多演示和手把手解决学员操作上的问题。中级班最理想的学员,或者说能从中级班中获益最大、学个痛快的,是那些已经做过一段时间量化计算,并且对量化计算真正有兴趣的学员。很多中级班讲的东西确实也得是亲自做过量化的人才懂得多么重要,听了才会豁然开朗或产生强烈共鸣,而此时零基础的学员可能都完全搞不懂台上在讲什么。
总之,零基础的学员和基础薄弱的学员一定要从初级班起步,能起飞了,再去考虑飞得更高更远,循序渐进,学习效率才是最高的。知识绝不是一下子学得越多越好,否则就像狼吞虎咽时会被噎住。有很多东西虽然重要,比如静态相关是计算电子结构复杂体系必知的概念,自旋污染对开壳层体系也是很重要的概念,配分函数是彻底搞明白热力学量是怎么计算出来的关键,但像这些概念,一下子灌输给零基础和基础薄弱的学员不仅没让他们学到更多,反倒容易使他们被搞糊涂,最麻烦的是容易把不相关的概念瞎联想到一起进而产生出各种莫名其妙的问题,限于他们的当前知识基础和理解能力讲又很难讲给他们明白。所以参加培训一定要选择适合自己当前水平的,否则只会事倍功半。
如果你不知道目前的水平是否适合参加中级班,可以做这个自测题:《初级量子化学培训自测题-基组部分》(http://bbs.keinsci.com/thread-5426-1-1.html)。如果大多数都不会,说明还没达到能参加中级班的水平。
中级量化班讲授的详细内容介绍见:http://www.keinsci.com/workshop/KBQC_content.html。
高级量化班:
高级量化班是给那些能力比较强,在完整消化完中级班的知识后还想再上一层楼,用更高级的手段研究更复杂高深问题的人开设的。高级量化班和中级班没有任何重复,讲的都是高级理论方法、小众向内容,比如CASSCF、多参考方法(NEVPT2、MRCI等)、从头算动力学、显式相关计算、低标度耦合簇方法、CCSDT(Q)和EOM-CCSDT等超高精度耦合簇计算、spin-flip TDDFT方法、全电子量子化学计算和旋轨耦合、约束性DFT计算、交换-相关泛函的调控、巨快速计算大体系光谱的sTDA方法、振动电子光谱的计算、双光子吸收的计算、搜索势能面极小能量交叉点、磷光速率的计算、内转换和系间窜越速率的计算,等等。而且会非常详细、完整讲解特别强大的ORCA量子化学程序的使用、极高效的xtb程序的使用,还介绍GAMESS-US、MRCC、NWChem、Dalton等初级和中级班没涉及的量化程序的部分功能的使用。注意,参加高级班之前必须先有中级班程度的知识基础,高级班讲授的内容假定学员已经参加过了中级班,或者至少具有同等程度的知识水平。如果初级、中级、高级班的内容都完整学完了,那么在量子化学方面的知识可以达到近乎完备的程度,是真正的高手了!
高级量化班讲授的详细内容介绍见:http://www.keinsci.com/workshop/KAQC_content.html。
简单归纳一下,三个量化培训班之间是以下关系:
此外,北京科音每年都举办“量子化学波函数分析与Multiwfn程序培训班”,为时5天(另赠送14小时的补充视频,故相当于5+1.5天)。初级/中级/高级量化班侧重于讲授怎么算,而波函数分析培训则重在讲授怎么分析。波函数分析用处极大,对于应用性量子化学研究是几乎必不可少的!充分掌握波函数分析,明显能让研究的分析、讨论部分深入、充实得多,令文章显著增光添彩,大幅提升发表的文章的档次。笔者开发的Multiwfn是实现波函数分析特别强大的程序,被Nature、Science、JACS、Angew、PNAS等诸多顶尖刊物的文章使用,目前用户已遍90多个国家,截止到2024年已被引用超过25000次!Multiwfn的一些相关介绍见《Multiwfn FAQ》(http://sobereva.com/452)和《Multiwfn入门tips》(http://sobereva.com/167)。Multiwfn程序不光是用于波函数分析上,对于最浅层次的量化研究也特别有用,比如可以快速方便地看轨道、绘制各种光谱图,所以在量化初级、中级班当中也会穿插着使用,但全面、系统、深入详细的讲解还是留在波函数分析培训班里。波函数分析与Multiwfn程序培训班的详细讲授内容见 http://www.keinsci.com/workshop/WFN_content.html。
北京科音每年还举办CP2K第一性原理计算培训班,介绍见http://www.keinsci.com/workshop/KFP_content.html。量子化学和第一性原理都是使用基于量子力学的理论方法来研究化学问题,但前者主要研究的是孤立体系(分子、团簇),而后者主要研究的是周期性体系(特别是固体与表面),这两个领域的研究对象是完全互补的,适用的程序、涉及的概念也有极大的不同。参加量子化学培训班后再参加CP2K第一性原理计算培训班,将使你的研究能力、眼界扩展一倍以上,因此非常推荐有机会参加。
北京科音每年也举办分子动力学与GROMACS培训班。分子动力学和量化都是计算化学的最重要组成的部分,搞量化的人,即便是不专门做动力学的,参加这个班开阔眼界、了解动力学计算的基本过程和基本思想,对未来也是很有好处的,能激发灵感、深化对计算化学的理解,偶尔碰到必须通过动力学才能解决的问题也不发愁了。北京科音分子动力学与GROMACS培训班的详细讲授内容见 http://www.keinsci.com/workshop/KGMX_content.html。
经常有人咨询北京科音办的培训的一些问题,常见的在此进行了统一解答:《北京科音办的培训班FAQ》(http://bbs.keinsci.com/thread-5098-1-1.html),可以看看是否其中也有自己想问的问题。如果你是微信用户,担心忘看通知而错过北京科音的培训,那么十分建议订阅北京科音微信公众号,只要搜索"北京科音"即可找到。
大家务必注意,如今社会上虚假、骗钱、山寨培训多如牛毛,参加了这种培训,不仅学不到什么东西,还白花了时间和金钱,所以一定要谨慎辨别。强烈建议参考:《辨别虚假坑钱科研培训的关键九点》(http://sobereva.com/339)。社会上还有些虽然是正规机构办的培训,但是质量真是奇差无比,和山寨培训本质没有任何两样,参加这种培训没有任何益处,白费时间白费钱,例如看此贴里几位学员曝光的一个培训真相:http://bbs.keinsci.com/thread-20785-1-1.html。参加某些机构的培训甚至还可能学到一大堆错误的东西,看过这个帖子你就能清楚体会到我为什么这么说了:http://bbs.keinsci.com/thread-21551-1-1.html。
二 自学
如果因为种种原因,没机会参加北京科音办的培训,那只能自学。周围的人,即便有懂行的,基本也没可能会系统地教你,必须靠自己摸索。虽然这样入门速度比参加培训慢得多得多得多,但只要按照如下我说的,至少入门过程中不会走向歪门邪道、被误导。(1) 看Levine的Quantum Chemistry。市面上有影印版。这本书对于专门搞量化理论的人来说只是初级水平,而对搞应用性计算的人,看完此书后基础知识方面勉强够了。此书写得很好,很系统,公式推导很易懂(只要高数能及格的人看着就没压力),量化常见概念也都有解释。但是,不要以为光读了这本书就算入门了,这本书不告诉你怎么结合Gaussian等主流程序算实际问题,不会用量化解决基本问题就还称不上入门。另外,此书对于广大量子化学初学者入门的一个很大问题就是铺垫太多,前半本书都在讲量子力学,可能都看累了、完全失去兴趣了,结果连HF还没明白是什么意思。所以,这本书,如果你有充裕的学习时间,慢慢从头到尾看完是很好的,但如果没那么多时间,别试图把此书都从头到尾细致啃完、习题都做了一遍才开始算实际问题。
(2) 看Exploring Chemistry With Electronic Structure Methods。这是Gaussian官方的书,专门讲怎么用Gaussian算各种具体实际化学问题,给了很多例子,对于初学者学习Gaussian使用实属难得。此书虽然也讲了点理论,但讲得很少,即便对于应用性量化研究的人也完全不够用。一定要看第二版,切勿看最新的第三版。第三版相对于第二版彻底重写,但是反倒更差,里面有很多误导性的东西,不少地方数据错误或缺乏逻辑,而且有用的例子更少,废话(而且是那种真正意义上的废话并不是有助于初学者理解问题的废话)还特别多。第二版bug不多,写得质量不错,可惜年代久远(1996年),书里用的计算级别在现在看来大多已经太低或过时了,但至少没有误人子弟或者科学、逻辑方面的问题。要注意,切勿以为把exploring这本书看了、都搞明白了,Gaussian就能用得顺利、游刃有余了,大多数在实际计算中必知的关键性的知识、技巧,在这书里没有强调或者根本都没提及,不懂这些的话在实际研究中经常会被卡住或者犯错,比如究竟应该如何合理有效地解决不收敛以及不该有的虚频,这些重要内容有的会在笔者的博文中介绍,但全面学习的机会只有在科音的培训中。
值得一说的是,如果你已经参加过科音的中级量化培训班了,就完全没必要再看Exploring这本书了,因为培训中讲的内容比书里全面丰富得太多太多了,制作幻灯片的时候笔者也已经把无数本计算化学书籍里有价值的内容都提炼并体现到其中了。但Levine那本书,参加过中级班之后只要有时间一定要系统地看看,可以加深对量子力学方面的认识,而书里有的量化部分的内容在中级班里都有,看的时候也可以顺便再回忆巩固一下。
(3) 看笔者的blog思想家公社的门口(http://sobereva.com)里头界面右边的量子化学分类里面的文章。笔者写过几百篇和量子化学相关的博文,写得都十分用心(笔者用写论文的态度写帖子),涵盖面非常广,讲解非常细致。有很多博文对初学者很有益,但也有的博文可能初学者只能看懂部分或完全看不懂。
注意:此博客的文章皆为笔者(sobereva)原创,而且笔者经常更新过往文章的内容以令文章与时俱进。此博客有大量文章,被很多人未经授权胡乱转载到了别处去,笔者表示谴责。千万别在sobereva.com和bbs.keinsci.com以外的地方(比如小*虫、他人的博客、百度文库)看笔者的文章!因为其它地方看到的都是老旧的而且格式/图片被转载者搞得乱七八糟的版本!(这两个地方首页都有搜索框,想找笔者写的某篇文章,只要一搜就有)
(4) 在思想家公社QQ群(群号看http://sobereva.com上方的公告)里关注大家的讨论。这是笔者建立并管理的人气极高、专业性特别强的计算化学讨论群,1号、2号和3号群成员总计达9000人(长期爆满。已经踢过的长期不发言的人加起来都已经一万人以上了),加入这个群对新人们来说算是找到组织了。每天群里都会有大量讨论,低、中、高档次问题都有。加入之后一定不要仅在自己提问的时候才看群消息,一定要每天都看群里的讨论,能看懂多少看多少。虽然这种方式学习并不系统,但经过长时间熏陶,水平能提高不少。
(5) 计算化学公社论坛(http://bbs.keinsci.com)。这是北京科音自然科学研究中心旗下的高水平计算化学论坛,由笔者创立和管理。此论坛是人气极高的计算化学交流论坛,甚至很多外国人都通过Google翻译看此论坛上的帖子。此论坛有高度的学术纯粹性,并且坚决抵制水贴,笔者每天都花大量时间精力回复论坛上的巨量问题和打理论坛。这个论坛从2014年10月开始运行,已积攒了海量极具价值的讨论。计算化学研究者应保持每天都看这个论坛新帖的习惯,量化版以往的帖子也都强烈建议在有空的时候一点点看完,能看懂多少看懂多少,特别是置顶的“简单量化问题答疑专贴”(http://bbs.keinsci.com/thread-806-1-1.html),对初学者更是非常有益。另外,碰到计算化学问题时,在发帖求助前,应当优先想到使用计算化学公社论坛首页Google框进行搜索。计算化学公社论坛已有的帖子是巨大的计算化学资源宝库,不是很冷门的问题大多都能从中搜到有关的帖子
在计算化学公社和思想家公社QQ群里每天笔者都会回复大量问题,日均好几十个。在这两个地方看到的讨论可以很放心,因为如果有人给出错误的回复,笔者只要看见一定会将之订正以免误导初学者。作为初学者,一定一定一定不要轻信网上其它任何地方的乱七八糟的和量子化学、计算化学相关的讨论,尤其是中文的,那些讨论中很多都是错的、误人子弟的,很多人自己还是菜鸟程度就随便回复别人问题,各种以讹传讹。那些缺乏高手把关的地方的讨论,初学者看多了不仅学不到有用的,反倒会对理论、概念产生错误的理解,在程序使用上学一身坏毛病,各种瞎用关键词。
不仅是网上的中文讨论不可轻信,网上的各种乱七八糟的中文教程、幻灯片之类的资料往往也都是包含各种错误,甚至有的满篇都是错的。入门阶段,不去看那些是最安全的(至少等你已经有一定水平了,有了基本分辨能力了再去看)。在计算化学公社论坛里下的资料都是放心、可信的,我是不会让有误人子弟的资料出现在论坛里的。
另外,市面上有不少中文的量化、计算化学的书。市面上所有这种书我基本都买过一本,买来不是为了阅读,就是单纯想看看书写得怎么样。整体感觉中文的这类书分三种:(1)照本宣科型。这类书的作者往往自己量化水平都没多深就写书,东抄西抄,根本没自己的东西,内容也都是老一套,大同小异,从量子力学开始讲,讲一大堆,然后讲HF、微扰等等。和主流、实际计算相关的东西在这种书里根本找不到。这种书根本别看,纯属浪费时间,也就适合在高校里当那种走形式的量化课的教材用。你如果真想极为系统地从量子力学开始慢慢学起直接看Levine的书就完了,何必看这些低水平的书籍。(2)有水平的理论书籍。比如徐的三大本就是这种。这种书有的号称是给新人看的,但实际上是开玩笑,初学者看了只会云里雾里、晕头转向,对量子化学产生无限的厌恶和恐惧。(3)实例教程型。有些书里讲一点基础理论,同时给了一些计算化学的例子,看似很适合初学者,有点exploring那书的感觉,但写得真是差远了。这些书理论部分就那么一丁点还写得不明不白,内容很不系统、全面,例子和练习给得很少而且往往也不怎么样,对初学者起不到由浅入深的启发、引导性,有的书里的例子甚至直接就是从exploring那本书里抄来的!(而科音的量化培训里面的例子、练习都是很用心设计的,都是原创的)有的这类书更是错误百出,尤其是讲基组的部分,可谓是重灾区,几乎就没多少中文书这里讲得是完全对的。我之前给一个初学者回答问题时发现,他看的某中文书居然把Pople基组加弥散和加极化函数的方式都搞混了。
简而言之,量化初学者别看市面上任何中文量化书,至少是目前来说,是真的没有适合初学者入门的。
顺带一提,一些初学者,明明四六级考得分不低,却在科研上畏惧英文,死活不愿意看英文经典的资料,非要看一些很烂的中文的资料。实际上,看中文资料,虽然语言理解上障碍更低,看似更容易阅读,但中文资料质量和英文的相比普遍相差甚远,在理解内容上需要突破的障碍、花得时间要多得多!所以,初学者非要看中文资料的这种做法是绝对划不来的!学术性的英文资料句式就那么些,生词也没多少,多看看很快就掌握那些常见生词了,阅读难度哪有四六级阅读理解高。更何况,发表英文论文,不得写成英文?用计算化学程序,不得看英文的输出和手册?不锻炼科技方面的英文水平,在搞科研上会受到极大的阻力。这个坎是早晚必须要克服(除非你的研究最终目的就是水一两篇中文文章勉强毕业)。
下面说一些其它值得说的问题:
前面说过,量化入门光学理论绝对不行,一定要结合具体程序算算实际问题。第一个用的量化程序,在我来看一定是Gaussian才行。Gaussian的输入文件是所有主流量化程序里最简单的,功能是最全面的,常用的功能运算速度都很好,完全可以满足95%以上的应用性量化研究者的需要,地位如同量化界的Windows。而ORCA、GAMESS-US、molcas之类那些更复杂、更学术的量化程序则类似于Linux。让一个鼠标都没摸过的人一上来就用Linux哪行?Gaussian都不会用的初学者去学那些输入文件更难写、程序结构更复杂、对使用者理论知识要求更高的程序,要么一点也学不懂,要么花了很大力气才只会用那些程序算很简单的问题,而这些时间如果用在学Gaussian上,早就玩得很转,能开展实际工作了。而且,其它量化程序的用户数目远远少于Gaussian,相关的学习资源少得多,遇到问题得到别人回答的几率也低得多。目前量化程序是什么格局,从此文可以了解:《2018年度计算化学公社杯最常用的量子化学程序和DFT泛函投票结果统计》(http://sobereva.com/420)、《2021年计算化学公社论坛“你最常用的计算化学程序和DFT泛函”投票结果统计》(http://sobereva.com/599)、《2024年计算化学公社举办的计算化学程序和DFT泛函的流行程度投票结果》(http://sobereva.com/706),文中也包括我对目前一些量化程序的评价。值得一提的是,目前市场上有几种骗傻子的,带图形界面的商业味特别浓的量化程序,如果有代理商向你推销,大家千万别买!那几个程序功能又弱、又不灵活,还卖得巨贵(比Gaussian贵几倍甚至更多),根本不值那价钱,就仗着有个凑凑合合的图形界面忽悠想做量化计算的外行购买,特别黑心。用那种程序容易被搞量化的同行瞧不起。
很多初学者不注重系统性学习知识(参加培训或看基础性的书),就知道看文献,以为文献看多了就会搞研究了,这是大错特错!文献根本不可能由浅入深系统地对知识进行讲解,那些讲解也不可能作为文章发表。看一大堆文献得到的只是一堆零零散散的知识碎片,没有基础性的知识根本不可能理解它们、将它们正确关联到一起形成正确的知识体系。虽然也有不少计算方法的综述类文献,但也不可能从最基本的、考虑到零基础的人的接受能力和知识背景去从头细致地慢慢讲。起步阶段的人,我建议少看文献多学基础知识,等入了门有能力上手做计算了,再看一部分和实际研究主题有关的文献和比较浅显易懂的理论方法类的综述。类似地,光是整天看网上的讨论,也不可能获得系统性的知识,只能一知半解地了解一些零散的概念,学一些零碎的计算技巧。关注计算化学公社和思想家公社QQ群里的讨论、答疑对提高水平很有益,但终究只能起到补充作用,搞量化计算所需的关键性的基础必须要通过前面提过的正规途径来学。
很多初学者一个特别坏的毛病是唯文献是从、文献里说什么就是什么,甚至都感觉到文献里的做法有不合理之处,也不假思索地盲从文献的做法。正所谓尽信书不如无书,尽信文献不如无文献。很多文献里的东西都是错的,一些明显的错误以及不合理的做法甚至于出现在IF很高的期刊中(连JACS也一样)。牵扯到理论计算的文章那么多,哪可能所有文章的作者、审稿人同时都是相应领域的专家,而且审稿人还都认真负责仔细评审?我特别反感的一个情况就是,有些初学者的计算方式明显是错的、用的计算级别明显是不合理的,给他指正,似乎还不服气,告诉我说“xxx文献里就是那么算的”。那些国际知名的很经典、很有名的基础性书籍或者专著,特别是那些出了好几版的书、作者在领域内名气很大水平很高的人写的书,存在错误的几率较低,初学者看这些书的时候即便盲信往往也没太大问题(但如果书的年代较早,里面有些观点是过时的。尤其是涉及到计算级别选择的问题,绝对不要看10年前的书)。而文献里,特别是那些水准不明的人写的,有错的几率那可太高了,盲目效仿文献注定要吃大亏。一个典型例子就是这篇JACS文章:https://pubs.acs.org/doi/10.1021/jacs.8b04642,对一个小分子居然用MP2/STO-3G扫描势能面,用HF/3-21G找过渡态,稍微有点最基本常识的量子化学研究者都知道这是极度荒诞的,简直是反面教材。初学这要是效仿这篇文章用的计算级别算自己的体系,碰到稍微懂一点量子化学的审稿人,文章都会100%被打回去。别人文章发出去了那纯粹是因为人家运气超级好,多个审稿人里恰好一个量化领域的专业审稿人都没有,或者审稿人正巧是其熟人。
要知道搞量化的分为两派,一派是搞理论方法、算法的,由于难度高、门槛高,所以只占很小部分,而绝大部分都是搞应用性计算的。这两类研究者所需的理论知识层次是截然不同的。搞理论方法的需要掌握很深的知识,还必须会编程,搞量化应用的人在入门时切不能向这些人看齐,看的书不能是给这些人写的书。
量子化学学习和研究过程中需要充分利用搜索引擎。搜研究文章一定要用Google学术,搜其它类型的科研相关的东西一定要用Google搜索,Google可以说是笔者科研过程中的左膀右臂。这里重点强调一点:千万别用bai*!bai*是搜什么的?那是用来搜娱乐八卦、有害、坑爹、低级趣味信息的,这么low的东西怎么能用来搜索学术信息?在笔者来看,bai*搜索是几乎最下作、最没素质、最唯利是图的公司搞的最糟糕的搜索引擎,搜索出的信息质量极差,对你最有价值的信息多数情况搜不到,反倒是垃圾、无意义、低水平的信息的权重贼老高,占满了搜索呈现页面,往往你得翻好几页才能找到点靠谱的信息,笔者对此体会太深了。而且,除了计算化学公社论坛、思想家公社blog等地方外,富有价值的量化方面的资料大部分是外文的,bai*这种东西连中文的学术方面的信息都搜不利落,用它试图搜索出对你有用的外文资料那更是天方夜谭。根本甭指望bai*对你的科研工作能有什么帮助,它纯粹是在浪费你的时间,越用bai*科研水平越低,而勤用Google才能令你登山更高台阶。千万别在高水平的讨论群和论坛说什么“我用bai*搜了xxx”、“bai*一下”,这一定会遭被群嘲。搜索学术信息,bai*至多至多给本科生用,而研究生及以上的人还用bai*这么弱智的东西只会被同行笑话。bai*百科那种水准低劣的东西更是千万别信,对科研工作者只会产生严重误导;而相对的,英文wiki上的信息则质量整体较高,多数是真正内行人用心写的,很少有严重误导性的内容,因此看百科必须去英文wiki上看(中文wiki质量和英文wiki比还差一大截)。另外,还有那个bai*学术,就是个Google学术的赤果果的山寨品,做得奇烂,品质和Google学术比差了十万八千里,对科研工作有害无益。当然,由于特殊情况,在有的地区Google搜索和Google学术是没法直接访问的,但是作为科研工作者,总得具备点上网知识吧。更别说什么“我没法用Google”、“Google打不开”于是就自取其辱,弃明投暗而改用bai*。
关于编程。有些初学者误以为搞量化还必须得学编程,这明显不对。对于搞应用性研究的人,至少在入门阶段,完全用不着编程,懂理论常识会用Gaussian算常见问题即可。但是鼓励有空的时候学学shell编程写脚本,这对研究大批量分子、或者大量同类问题的时候能节省巨额操作上的劳力,而且提取数据这种事靠脚本来做还免得人工处理时候犯糊涂给搞错。shell编程其实很简单,学几个小时就能解决很大问题,典型例子可以看看比如《使用Gaussian时的几个实用脚本和命令》(http://sobereva.com/258),在《详谈Multiwfn的命令行方式运行和批量运行的方法》(http://sobereva.com/612)里有脚本编写深入浅出的相关知识介绍。
做量子化学计算终究还是要有一些基本化学直觉和高等化学常识的,但是有很多人本科不是化学出身的,这些人如果以后长期做量化研究,我觉得最好还是找机会补一补化学系本科期间会学到的最重要的知识,这样更容易更有效率地获得更有意义的研究结果。我觉得值得补的是:普通化学、结构化学、无机化学、有机化学、物理化学、生物化学、仪器分析。其中普通化学和结构化学是必须看的,其它的根据实际研究需要来选择性地看,并不需要看得精通,也不必做题,只要懂得基本概念就行了。至于看什么书,普通化学一定看北大的那本《普通化学原理》,结构化学一定看周公度的《结构化学基础》。其它的看什么无所谓,不用看太深的,个人认为无机化学看吴国庆写的那本就不错。有机化学不用看邢其毅那种偏深的,找本比较浅的看就够了,比如我本科时候用的是尹冬冬写的有机化学就还可以。物理化学书里最经典的是Atkin或Levine写的,国内有影印版,内容广度非常高(国外的物理化学和国内的物理化学不同,前者把结构化学和其它一些化学分支的部分知识都纳入了),如果能通篇看下来最好,但非常厚,大抵很多人也没时间看完,时间有限的人自己在书店里随便找一本感觉写得清楚易懂的就行。
初学者少不了一堆问题要问,但是很多初学者在提问时候习惯、方式很不好,甚至造成回复者的反感。关于提问要注意的内容在此文都说了,强烈建议看看:《在网上求助计算化学问题时的注意事项》(http://sobereva.com/79)和《在网上求助计算化学问题的时候必须把问题描述得详细、具体、准确、清楚》(http://sobereva.com/620)。其中我尤为想强调的有几点:(1)提问时候必须礼貌 (2)一定不要用大字号,这和公共场合扯着嗓门喊话一样没素质 (3)提问时候一定要把问题阐述清楚、准确,不要试探性地提问 (4)能自己解决的就一定不要问,好好看手册、用Google搜索,否则独立解决问题的能力很难有长进。
有些人可能自己懒得学、懒得算,或者由于计算经验不够需要他人帮助,或者没有服务器,因而想找他人代算。一定要注意,随随便便就找代算,被坑惨的几率超过90%!不仅可能白花上万,还极可能导致文章最终被撤稿!关于这点我专门写了篇文章,强烈建议一读:《谈谈我对计算化学代算的看法》(http://sobereva.com/505)。如果实在自己不会算,强烈建议找内行合作,而绝对不是花钱去社会上找人代算!如果就是要找代算,也极其强烈推荐通过北京科音计算化学培训班(见http://www.keinsci.com/workshop.html)系统学学相关知识,哪怕不想花时间学具体操作也至少要具备相关素养!否则被黑心的代算严重忽悠、坑上万块钱都浑然不知,而且也不知道怎么正确、有效地和靠谱的代算交流、提出计算要求和理解计算结果。
老有些初学者喜欢自己做测试,通过测试选择在计算时最恰当的方法,然而这种行为往往都是没有丝毫意义的无用功,在内行人眼里还会被笑话。关于这点我专门写过一个博文《谈谈量子化学研究中什么样的benchmark才有意义》(http://sobereva.com/554),非常建议仔细看看。
如今有一些初学者,妄图凭借ChatGPT、Grok等生成式AI的回答来学习计算化学,实际上以这种方式来学习很不靠谱(尤其是对于国内的AI来说)。这些AI输出的和计算化学有关的文字经常错误百出(来自于我的大量亲自测试),凡是内行人都能看出那些回答净是在一本正经的胡说八道。没有辨识能力的初学者看这种东西来学完全就是给自己喂毒药,明明自己都被带偏了却还觉得AI说得头头是道、以为掌握了学习的法宝。一定要明白生成式AI的长处和局限性是什么。训练AI模型的可靠的信息越充分,AI返回来的信息才可能越靠谱,而计算化学是非常专业且小众的东西(远不是本科层次的数理化可比的),互联网上的资源中能有效训练AI模型的信息相当有限(而且网上还净是错误的跟计算化学有关的文字,严重污染了AI的训练),再加上计算化学问题牵扯很多复杂的知识和逻辑,主流的AI模型自己在计算化学方面还是个糊涂虫(比如把cc-pVDZ说成是高精度基组、色散校正说成弥散校正...),跟它学完全是瞎绕弯路,还会被它给的各种错误的信息先入为主地占用了大脑,有害无益。做计算化学的也不是不能用AI,用的前提是你已经有了足够的知识基础,并懂得自己去查阅专业资料(像样的文献,以及诸如计算化学公社论坛上和我的博客中的靠谱的相关文字),从而有能力判断AI给你返回的信息有无参考价值、检验给出的说法是否合理。PS:公社QQ群里讨论时我听闻有初学者提到网上已经有一些视频讲怎么用AI学计算化学、动力学模拟,我对于制作视频的人在这方面是否真的已达到了入门水平都深表怀疑,这种视频我看也就是博人眼球罢了,撑死了也就是学一些最最基本的皮毛程度的概念,诸如什么叫势能面、分子力场的基本概念等;而靠它来学稍微深一点的知识,以及计算化学程序的正确使用(例如如何写超过hello world!难度的输入文件),那根本没戏。而且被AI带偏了还会导致你做大量毫无意义的计算、是内行人眼的胡算瞎算,白白浪费巨量的宝贵时间。比如最一开始AI建议你用的几何优化的方式就不对,导致后面你做的各种光谱计算、热力学数据计算、波函数分析等数据也全都泡汤了,如果还是计算耗时很高的大体系,被AI坑几次说不定你就要考虑延期毕业了。如果你还是用的超算算的,还要承担大量经济损失,说不定几千乃至几万块钱就被白白烧没了。我个人强烈建议读者尽可能参加北京科音的计算化学培训以正确方式入门和上手计算,到时候就算你用AI做为助手,只要先彻底搞懂了培训里很全面、系统系统讲的知识,也能分辨AI返回来的信息哪些不靠谱而哪些可以作为大致参考。
强烈建议好好看看此文!《谈谈计算化学新人使用生成式AI需要注意的问题与合理使用AI的方式》(http://sobereva.com/772)。
附:学习量子化学计算必看的博文
前面提到过,自学量子化学要大量看http://sobereva.com里的博文,但是我知道,对新人来说,把那里面所有量化相关文章在短时间内看完是不现实的,因此这里我把其中对量化计算初学者最最最重要、非看不可的博文整理了出来,缺乏文中的知识的话很难不犯低级错误地开展计算。注意这些博文只是涉及各种零碎的主题,零基础初学者光是看这些是无法从头系统性学习的,一次性系统地学明白的又好又快的途径是参加前述的北京科音的量子化学培训。
• 必须看:
Gaussian的安装方法及运行时的相关问题 http://sobereva.com/439
辨析计算化学中的任务类型和理论方法 http://sobereva.com/680
简谈量子化学计算中DFT泛函的选择 http://sobereva.com/272
谈谈量子化学中基组的选择 http://sobereva.com/336
谈谈赝势基组的选用 http://sobereva.com/373
谈谈弥散函数和“月份”基组 http://sobereva.com/119
详解Gaussian中混合基组、自定义基组和赝势基组的输入 http://sobereva.com/60
谈谈该从Gaussian输出文件中的什么地方读电子能量 http://sobereva.com/488
解决SCF不收敛问题的方法 http://sobereva.com/61
量子化学计算中帮助几何优化收敛的常用方法 http://sobereva.com/164
常见的多余的和被滥用的Gaussian关键词 http://sobereva.com/331
使用Multiwfn观看分子轨道 http://sobereva.com/269
在Gaussian中计算IRC的方法和常见问题 http://sobereva.com/400
谈谈如何又好又快地计算NMR化学位移 http://sobereva.com/354
使用Multiwfn绘制NMR谱 http://sobereva.com/565
revTPSS泛函结合pcSseg-1基组是计算NMR很好的选择 http://sobereva.com/623
谈谈隐式溶剂模型下溶解自由能和体系自由能的计算 http://sobereva.com/327
Gaussian中用TDDFT计算激发态和吸收、荧光、磷光光谱的方法 http://sobereva.com/314
Gaussian中几何优化收敛后Freq时出现NO或虚频的原因和解决方法 http://sobereva.com/278
使用Multiwfn绘制红外、拉曼、UV-Vis、ECD、VCD和ROA光谱图 http://sobereva.com/224
浅谈为什么优化和振动分析不需要用大基组 http://sobereva.com/387
使用Shermo结合量子化学程序计算分子的各种热力学数据示例 http://sobereva.com/552
谈谈谐振频率校正因子 http://sobereva.com/221
在Gaussian中做限制性优化的方法 http://sobereva.com/404
简谈Gaussian里找过渡态的关键词opt=TS和QST2、3 http://sobereva.com/460
详谈使用Gaussian做势能面扫描 http://sobereva.com/474
谈谈原子间是否成键的判断问题 http://sobereva.com/414
正确地认识分子的能隙(gap)、HOMO和LUMO http://sobereva.com/543
谈谈重复不出来计算化学文献里的数据的可能原因 http://sobereva.com/678
充分运用波函数分析的理论计算文章的模板 http://sobereva.com/764
• 看完以上内容之后应当继续看以下内容,对一般性研究都非常重要:
Multiwfn入门tips(http://sobereva.com/167)(Multiwfn是量子化学应用性研究离不开的重要程序)
Multiwfn FAQ http://sobereva.com/452
Multiwfn波函数分析程序的意义、功能与用途 http://sobereva.com/184
Multiwfn支持的电子激发分析方法一览 http://sobereva.com/437
Multiwfn支持的分析化学键的方法一览 http://sobereva.com/471
使用Gaussian时的几个实用脚本和命令 http://sobereva.com/258
谈谈Gaussian中的对称性与nosymm关键词的使用 http://sobereva.com/297
计算化学中的一些常见不良写法和用词 http://sobereva.com/298
量子化学研究中切换泛函应当注意的问题 http://sobereva.com/415
谈谈温度、压力、同位素设定对量子化学计算结果产生的影响 http://sobereva.com/423
Gaussian中有用的IOp一览 http://sobereva.com/93
Gaussian的Link、IOp与非标准计算路径 http://sobereva.com/57
过渡态、反应路径的计算方法及相关问题 http://sobereva.com/44
基于过渡态理论计算反应速率常数的Excel表格 http://sobereva.com/310
使用Multiwfn绘制构象权重平均的光谱 http://sobereva.com/383
乱谈激发态的计算方法 http://sobereva.com/265
根据Boltzmann分布计算分子不同构象所占比例 http://sobereva.com/165
谈谈轨道成份的计算方法 http://sobereva.com/131
谈谈片段组合波函数与自旋极化单重态 http://sobereva.com/82
各种后HF方法精度简单横测 http://sobereva.com/378
计算化学购机配置推荐 http://sobereva.com/444
写计算化学文章时引用理论方法、基组、程序时应注意的问题 http://sobereva.com/370
详谈Multiwfn产生ORCA量子化学程序的输入文件的功能 http://sobereva.com/490
2021年计算化学公社论坛“你最常用的计算化学程序和DFT泛函”投票结果统计 http://sobereva.com/599
2024年计算化学公社举办的计算化学程序和DFT泛函的流行程度投票结果 http://sobereva.com/706
谈谈如何通过势垒判断反应是否容易发生 http://sobereva.com/506
一键把所有gjf文件转成xyz文件、把所有Gaussian输出文件转成gjf文件的脚本 http://sobereva.com/530
Gaussian中非内置的理论方法和泛函的用法 http://sobereva.com/344
谈谈量子化学研究中什么样的benchmark才有意义 http://sobereva.com/554
实验测定分子结构的方法以及将实验结构用于量子化学计算需要注意的问题 http://sobereva.com/569
谈谈不同量子化学程序计算结果的差异问题 http://sobereva.com/573
使用Gaussian做镧系金属配合物的量子化学计算 http://sobereva.com/581
谈谈有小虚频时热力学量的计算 http://sobereva.com/699
谈谈怎么考察、计算、分析化学体系的电子密度 http://sobereva.com/715
化学体系的静电势的计算和分析方法汇总 http://sobereva.com/769
Multiwfn支持的预测化学体系反应位点和反应活性方法一览 http://sobereva.com/767
• 如果你需要研究弱相互作用,以下文章非常建议看
乱谈DFT-D http://sobereva.com/83
DFT-D色散校正的使用 http://sobereva.com/210
DFT-D4色散校正的简介与使用 http://sobereva.com/464
谈谈“计算时是否需要加DFT-D3色散校正?” http://sobereva.com/413
谈谈BSSE校正与Gaussian对它的处理 http://sobereva.com/46
Multiwfn支持的弱相互作用的分析方法概览 http://sobereva.com/252
使用sobEDA和sobEDAw方法做非常准确、快速、方便、普适的能量分解分析 http://sobereva.com/685
大体系弱相互作用计算的解决之道 http://sobereva.com/214
计算分子内氢键键能的几种方法 http://sobereva.com/522
透彻认识氢键本质、简单可靠地估计氢键强度:一篇2019年JCC上的重要研究文章介绍 http://sobereva.com/513
Angew. Chem.上发表了全面介绍各种共价和非共价相互作用可视化分析方法的综述 http://sobereva.com/746
一篇最全面介绍各种弱相互作用可视化分析方法的文章已发表! http://sobereva.com/667
谈谈分子间结合能的构成以及分解分析思想 http://sobereva.com/733
全面探究18碳环独特的分子间相互作用与pi-pi堆积特征 http://sobereva.com/572
使用量子化学程序基于簇模型计算金属表面吸附问题 http://sobereva.com/540
18碳环(cyclo[18]carbon)与石墨烯的相互作用:基于簇模型的研究一例 http://sobereva.com/615
要善用簇模型,不要盲目用ONIOM算蛋白质-小分子相互作用问题 http://sobereva.com/597
在ORCA中做counterpoise校正并计算分子间结合能的例子 http://sobereva.com/542
谈谈pi-pi相互作用 http://sobereva.com/737
• 如果你计算的分子有高度柔性,需要做构象搜索,或者研究分子复合物,需要做构型搜索,务必看
molclus主页:http://www.keinsci.com/research/molclus.html
Molclus FAQ http://sobereva.com/730
使用molclus程序做团簇构型搜索和分子构象搜索
http://bbs.keinsci.com/thread-577-1-1.html
gentor:扫描方式做分子构象搜索的便捷工具
http://bbs.keinsci.com/thread-2388-1-1.html
genmer:生成团簇初始构型结合molclus做团簇结构搜索的超便捷工具
http://bbs.keinsci.com/thread-2369-1-1.html
将Confab或Frog2与Molclus联用对有机体系做构象搜索
http://bbs.keinsci.com/thread-20063-1-1.html
使用Molclus结合xtb做的动力学模拟对瑞德西韦(Remdesivir)做构象搜索
http://bbs.keinsci.com/thread-16255-1-1.html