正确地认识分子的能隙(gap)、HOMO和LUMO
正确地认识分子的能隙(gap)、HOMO和LUMO
Correctly understanding energy gap, HOMO and LUMO of molecules
文/Sobereva@北京科音
First release: 2020-Mar-26 Last update: 2020-May-2
0 前言
笔者在QQ群(http://sobereva.com/QQrule.html)里、计算化学公社论坛(http://bbs.keinsci.com)里答疑时经常看到有计算化学初学者、理论计算外行对gap、HOMO、LUMO的理解存在严重误区,很多错误还存在各种文献中,长期以来以讹传讹,诸如什么“轨道能量差就是激发能”、“循环伏安法能测HOMO能级”等等。在此就写一篇文章,让缺乏理论化学知识的人能对分子的gap、HOMO、LUMO有个正确的认识,分清楚不同gap之间的区别,明白为什么轨道能量是不能实验测定的,最后再专门说说与本文主题有关的Koopmans定理。
1 分子体系gap的三种类型
首先要搞清楚,孤立体系,比如分子、团簇,有三种gap(能隙),绝对不能混淆:
(1) HOMO-LUMO gap:定义是E(LUMO)减去E(HOMO),显然肯定是个正值。平时说分子体系的gap的时候,如果没有前提,多数情况指的就是这种gap。
(2) Fundamental gap:定义是VIP减去VEA。其中VIP是vertical ionization potential的缩写,中文一般叫垂直电离能,定义是VIP = E(N-1) - E(N),这里E是电子能量,N是体系原本的电子数,N-1是电离掉一个电子之后的电子数。VEA是vertical electron affinity的缩写,文中叫垂直电子亲和能,定义是VEA = E(N) - E(N+1),N+1是指体系额外获得一个电子后的电子数。显然,fundamental gap等于E(N-1) + E(N+1) - 2*E(N)。
(3) Optical gap(光学gap):基态电子态通过吸收光子所能跃迁到的最低激发态对应的激发能。比如对于一般的有机染料分子,对应的就是S0态极小点结构下S0到S1态的垂直跃迁对应的激发能。由于跃迁禁阻而无法观测到的最低激发态不算,比如对于单重态基态体系,虽然T1态比S1还要低,但不能把S0与T1间的能量差叫做optical gap,因为这是对称禁阻的。
以上三种gap中,HOMO-LUMO gap是无法通过实验测定的,因为HOMO和LUMO根本就不是真实存在的东西,详见本文第4节,因此凡是见到有人说通过xxx实验测了HOMO-LUMO gap,一律都是胡说八道,或者错误地解释了实验。由于VIP和VEA都是可以实验测定的(气相VIP可以通过光电子谱得到,气相VEA可以通过电子附着光谱得到),因此fundamental gap是可以实验测定的。Optical gap通过UV-Vis光谱就可以直接测定,相当于波长最大的吸收峰位置(其实这不严格,因为有时相邻的激发态的吸收峰会显著相互叠加,导致实际观测到的一个峰可能对应不止一个激发态。另外还要考虑振动耦合问题,这影响峰位置,本文就不多说了,感兴趣者可以看《振动分辨的电子光谱的计算》http://sobereva.com/223。后文也忽略振动态的问题)。
所有以上三种gap都可以通过量子化学计算非常容易地得到。在基态极小点结构下,HOMO-LUMO gap用KS-DFT方法做个单点计算就有,optical gap用诸如TDDFT算一下激发态就有(见比如http://sobereva.com/314),算fundamental gap只需要把N+1、N、N-1态的单点能都算出来代入公式即可。
Optical gap比fundamental gap数值肯定要低,差值称为electron-hole pair binding energy。
老有初学者明明说的是分子体系,却用带隙(band gap)这个词,这是大错特错。周期性体系才有能带的概念,而分子体系根本都没有能带,何来带隙?
感兴趣的读者可以看看《mind the gap》(Mater. Horiz., 1, 17 (2014)),只有两页,专门说了这三种gap。文中还提到,fundamental gap是对于分子体系而言的,对于周期性体系来说,band gap=fundamental gap=transport gap,但和optical gap不同,因为也相差着electron-hole pair binding energy。周期性体系不能说HOMO-LUMO gap,因为HOMO和LUMO从其名字上就知道这本来就是对于分子(或者说孤立体系)而言的,对周期性体系可以类比的概念是LUCO(最低非占据晶体轨道)和HOCO(最高占据晶体轨道),分别对应于导带底和价带顶的位置,差值就是band gap。
2 HOMO-LUMO gap与fundamental/optical gap的关系
分子的HOMO-LUMO gap虽然和fundamental gap、optical gap不等价,但可以视为二者的近似,在一定程度上有正相关性,下面具体说一下。
Koopmans定理认为VIP≈-E(HOMO)、VEA≈-E(LUMO),因此VIP-VEA≈E(LUMO)-E(HOMO),即fundamental gap≈HOMO-LUMO gap。虽然二者有相关性,但是绝对不要期望在定量数值上能有多好的对应关系。因为VIP≈-E(HOMO)这个关系对于常用泛函来说误差是很大的,尤其是VEA≈-E(LUMO)这个关系极差,在本文第4节还会专门说。
不少搞实验化学的,以及不少仪器分析、有机化学之类的书籍里,认为LUMO与HOMO间的能量差就对应于最低电子激发能,即把HOMO-LUMO gap和optical gap划等号,这是大错特错!要明白,电子激发过程是从一个电子态(电子基态)到另一个电子态(电子激发态)的跃迁,而不是从一个占据轨道到一个非占据轨道的跃迁。电子态是真实存在的,而轨道只是虚构出来的概念。看过《电子激发任务中轨道跃迁贡献的计算》(http://sobereva.com/230)一文就知道,比如常用的算电子激发的TDDFT方法,把电子激发在数学上描述成各种各样形式的轨道跃迁的线性组合。虽然对于某些体系,通过查看TDDFT的单激发组态函数的系数会发现,能量最低的电子激发方式确实可以近乎100%地通过HOMO->LUMO的跃迁来描述,但也绝对不能把这俩轨道的能量差(即HOMO-LUMO gap)直接当做激发能,因为二者之间还相差激子结合能。这里引用笔者讲授的北京科音中级量子化学培训班(http://www.keinsci.com/KBQC)的幻灯片里的一页来说明

上图中的激子结合能绝对不能忽略,它导致HOMO-LUMO gap与TDDFT算的optical gap之间相差零点几eV是非常正常的。还要知道,最低电子激发即便有一对轨道贡献近乎100%,这对轨道也未必是HOMO和LUMO。比如反式偶氮苯,在基态极小点结构下用TD-PBE0/6-31G*下计算出来的S0→S1激发对应的是HOMO-1→LUMO跃迁,而S0→S2才对应于HOMO→LUMO跃迁,这是因为两种电子激发的激子结合能的不同导致激发能与轨道能量差不满足正相关性。另外,由于对称禁阻,或者恰好最低激发态的振子强度就是特别小,实验吸收谱上观测到的最大波长的峰位置可能并非是最低激发态,换句话说,最低激发态是个暗态(dark state),这种情况下显然就更甭指望optical gap能和HOMO-LUMO gap联系起来。
虽然HOMO-LUMO gap原理上与optical gap不等价,对于实际体系,从统计结果上看关系如何?在J. Chem. Inf. Model., 57, 1300 (2017)中,作者计算了2248895个小分子体系的最低激发能与HOMO-LUMO gap,得到下图

可见二者间虽然有正相关性,但是相关性比较弱,数据点分布相当分散。
在实际研究中,拿量子化学程序算出来的HOMO-LUMO gap去解释理论计算或者实验观测到的fundamental gap、optical gap的趋势,在一定程度上是可以的,但如果解释不好,也绝对不要强求,否则就是强词夺理了。而把HOMO-LUMO gap直接当做fundamental gap或optical gap来用,是绝对不能接受的!碰上稍微懂点理论计算的审稿人一定会挨批。
还要注意,计算的HOMO-LUMO gap受使用的DFT泛函影响非常大,通常是HF成份越高的泛函,算出来的HOMO-LUMO gap越大。从下图笔者计算的数据中可以清楚地看到这一点。
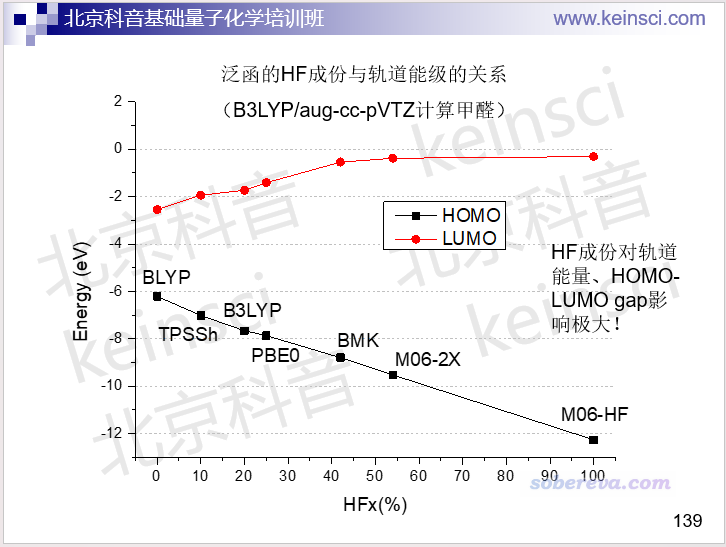
某种泛函算出来的HOMO-LUMO gap大小,和它计算出的fundamental gap或optical gap的准确性毫无必然关系。而且本身HOMO-LUMO gap也不是实验可测的,完全不能以这个量的大小判断泛函的好坏。
3 HOMO-LUMO gap的一些用处
HOMO-LUMO gap除了作为optical gap和fundamental gap的近似外,还有一些实际用处,在这里提一下。
寡聚物的HOMO-LUMO gap常被用于衡量分子的导电性。HOMO-LUMO gap越小,一般认为导电性越好。例如笔者和合作者在RSC Adv., 3, 25881 (2013)中研究了一系列寡聚物的能隙,如下所示,苯和萘以不同排列、不同比例进行组合,使体系展现出了不同的HOMO-LUMO gap。这体现出寡聚物像蛋白质一样,通过序列可以决定其功能性(控制导电性)

寡聚物的HOMO-LUMO gap和无限延展的聚合物的带隙存在密切联系,可以通过量子化学程序计算的HOMO-LUMO gap来外推出带隙。下图来自Acc. Chem. Res., 44, 14 (2011)
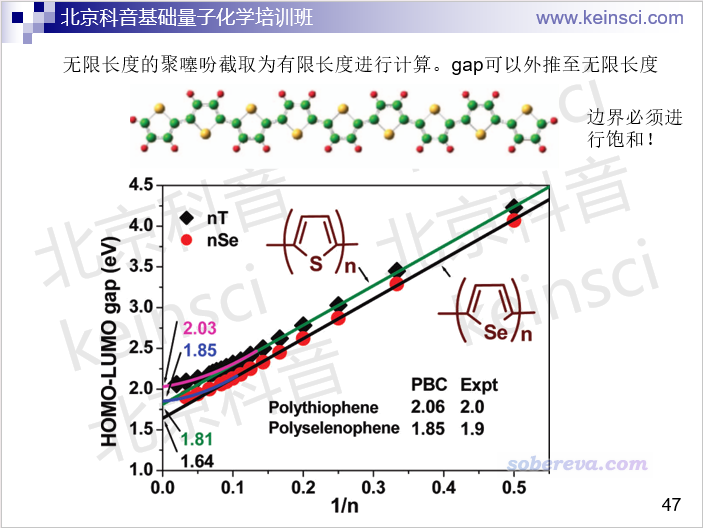
文中用B3LYP/6-31G*对不同单元数的噻吩寡聚物计算了HOMO-LUMO gap,并且外推到无穷长的数值,如图中粉字所示,结果为2.03 eV,这和用周期性计算聚噻吩得到的带隙2.06 eV非常相近。周期性体系的带隙是可以直接实验测的,实验值是2.0 eV,可见理论计算的和实验很相近。
这篇Acc. Chem. Res.文章和不少研究寡聚物的文章中都提到,B3LYP/6-31G*很适合算聚合物的导电性。另外,在J. Phys. Chem. Lett., 7, 1198 (2016)中,作者对于各种无机体系做了测试,发现B3PW91算带隙算得很理想,平均误差只有0.28 eV。由于B3LYP和B3PW91的特征相近,HF成份都是20%,因此此文的结论对于B3LYP也适用。虽然前面说了,分子体系的HOMO-LUMO gap是不可观测的,但考虑到它与带隙之间的关系,倘若非要说HOMO-LUMO gap拿什么泛函算比较合适,通常来说B3LYP或B3PW91是优先值得考虑的,对于算这个问题二者结果几乎一样,见比如RSC Adv., 3, 25881 (2013)文中图2的对比。
概念密度泛函是研究化学反应活性和位点的一套重要理论,详见《概念密度泛函综述和重要文献合集》(http://bbs.keinsci.com/thread-384-1-1.html)中的资料。在这个框架中有一个量叫软度(softness)。软度不是指体系的刚性大小,而是反映电子的活泼性、其分布的易变形程度。对于一系列类似体系(比如某些官能团不同、局部结构有差异,但主体特征一致),通常认为软度越大,分子越容易发生反应。软度的表达式为1=1/(VIP-VEA),可见它等价于fundamental gap的倒数;而在Koopmans近似下,软度直接等于HOMO-LUMO gap的倒数。这是为什么很多文章喜欢拿HOMO-LUMO gap说事,谁比较小就说谁的反应活性更高。这确实有一定依据,但一定要注意这种分析仅限于类似物,若用于对比两个特征相差甚巨的分子的反应活性,那纯属伪科学。
有很多文章拿HOMO-LUMO gap衡量分子稳定性,谁的HOMO-LUMO gap越小就说谁越不稳定。这种说法相当粗俗,总是被用来瞎讨论。要注意“稳定性”绝对不是一个简单的词。不稳定是指当前分子容易变成其它构型或物质,这对应于很多可能的过程,因此可以区分成化学稳定性、光稳定性、热稳定性等等,显然这绝对不是简简单单光靠一个HOMO-LUMO gap就能说明的。倘若是讨论化学稳定性,并且不考虑另一种反应物的特征,且对比的一批分子之间有足够相似性,那么拿HOMO-LUMO gap来说事还算说得过去。如果另一种反应物是明确的,并且你有能力用量子化学程序计算出反应势垒,若发现HOMO-LUMO gap和势垒有正相关性,那么值得在文章中说一说,但如果与势垒没有这种相关性,则体现出HOMO-LUMO gap把问题过度简单化了,应当摒弃这种分析(如果强行分析,可能会被审稿人批,届时更麻烦)。
HOMO-LUMO gap越小通常极化率也越大,这点也值得一提。这可以从完全态求和公式角度理解,详见《使用Multiwfn基于完全态求和(SOS)方法计算极化率和超极化率》(http://sobereva.com/232)。由极化率表达式可见,分母有一项是激发能,这和主导电子激发的两个轨道间的能量差有相关性。HOMO-LUMO gap越小暗示着占据轨道与非占据轨道的能量差整体越小,进而倾向于令激发能也越小,故而极化率倾向于越大。
搞实验的人很喜欢拿HOMO-LUMO gap说事,在这里告诫一下初学者,千万不要盲目听信已发表的文章里的HOMO-LUMO gap的分析,尽信文献不如无文献。文章中说得在理的可以吸取,而显得莫名其妙的分析、不合基本化学逻辑的讨论,则绝对不要轻信和效仿。如果以为已经发表的文章就都是正确的,那实在太天真了。
4 HOMO和LUMO能实验测定么?
在化学领域里“循环伏安法测定的HOMO、LUMO是xxx”这种描述随处可见,有些文章更是声称测量了轨道波函数并给出了图像,甚至还发到了某些人梦寐以求的顶级刊物上。这类文章以讹传讹程度之广令人发指,明显错误的东西反复出现在诸多研究者的视野里,令他们信以为真。这使得笔者在量子化学波函数分析与Multiwfn程序培训班(http://www.keinsci.com/WFN)一开始就给出了两页ppt以正视听:


分子轨道是不可测量的这一点,根本不需要什么复杂的理论知识就能明白。只要有理论化学最基本的常识,知道Fock和Kohn-Sham算符是什么,自然就明白分子轨道是理论上做了单电子近似才有的概念,是将分子波函数以简化方式求解才诞生的,显然是纯粹人为虚构出来的产物。Hartree-Fock、半经验、KS-DFT方法全都可以得到分子轨道,具体来说,分子轨道是人为定义的单电子有效哈密顿算符的本征函数的统称。既然分子轨道在现实当中都不存在,谈何实验观测?在原理上怎么可能实验测定?显然,那些号称实验上测定的分子轨道根本就不是分子轨道,而是对观测到的数据的错误理解和解释,将其错误地冠上了“分子轨道”的名字。
那些通过光电子实验给出了“分子轨道”图像的文章,实际上观测到的是物理意义严格的Dyson轨道,仔细看看上面幻灯片里的文章就会了解得更多。分子轨道只不过很多情况是Dyson轨道的近似罢了。具体来说,对于静态相关弱的体系,Dyson轨道与分子轨道定性一致;对于强相关体系(静态相关强的体系,如双自由基),Dyson轨道可能与分子轨道显著不符,往往需要写为多个分子轨道的线性组合才能定性描述出来。所以千万不要把Dyson轨道与分子轨道混淆。只有当完全忽略电子相关以及轨道的弛豫,即Koopmans定理所假设的情况,那么Dyson轨道才正好是HF方法算的分子轨道。
那些搞电化学的人通过循环伏安法测的既然不是HOMO、LUMO的能量,那测的究竟是什么?看下面我总结的关系图(引用的话请注明出处,即本文的网址)
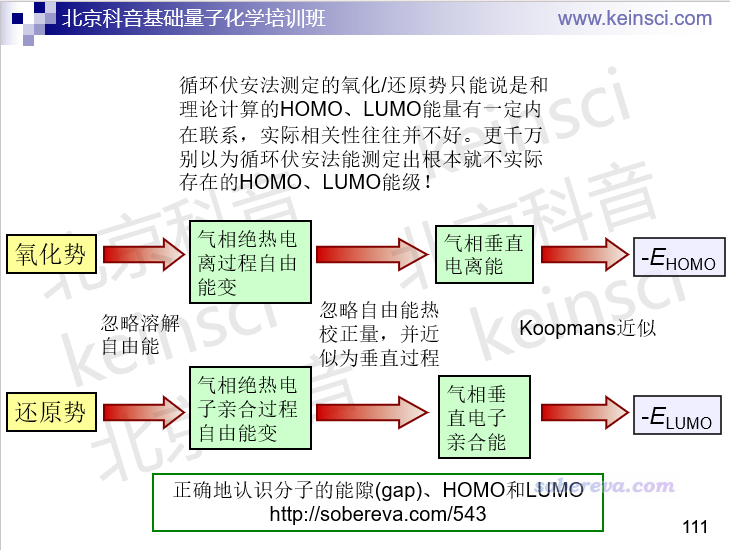
实际上,循环伏安法测的是溶液中的氧化势和还原势,这对应于电极反应过程的自由能变,离HOMO、LUMO能量隔着多达三层近似!显然把溶液中的氧化势和还原势直接分别当成HOMO和LUMO能量的负值是非常荒唐的,只能说存在正相关性而已,而这相关性还不怎么样。所以千万不要以为自己做量子化学计算算出来的前线轨道能量和循环伏安法实验数据对应得不好就以为自己算得不对。溶液中的氧化势和还原势都是可以通过量子化学方法(需要利用溶剂模型)计算出来的,这才和循环伏安法实验真正有可比性(但想对应得很好仍然很不容易,因为对于离子体系,现有的最好的隐式溶剂模型算的溶解自由能的误差也挺大,相关信息看http://sobereva.com/327)。
光电子谱可以测定将体系各层(不限于最外层)电子电离走对应的电离能。有人说光电子谱测定的就是分子轨道能量,实际上这也是错误的。前面已经说了-E(HOMO)只不过是最外层电子的电离能(第一VIP)的近似,对于其它层的电子也是一样。实际上,电离过程相当于N电子态跃迁到N-1电子态,由于电子相关作用,不能将这简单当成是一个轨道上的电子电离掉;而且哪怕是忽略掉电子相关问题,即N与N-1态的跃迁就相当于掉了一个轨道上的电子,但这个电子走了之后由于其它电子感受到的外势发生了变化,故其余的占据轨道会发生弛豫,也会导致体系能量进一步发生改变。显然将电离各层电子对应的电离能直接当做是各个分子轨道的能量的负值是明显错误的。
HOMO、LUMO不仅不可实验测定,而且也不能通过诸如CCSD(T)、NEVPT2等高精度量子化学方法来计算,这是因为这些方法不是像HF、KS-DFT那样是基于单电子近似框架的,方法本身也没有定义有效单电子哈密顿算符,因此根本无法产生分子轨道。有些人可能会问“我用Gaussian做完CCSD(T),不是给出了一批轨道能量么?”这实际上是做CCSD(T)电子相关计算之前做HF的那一步产生的HF分子轨道,根本不是什么“CCSD(T)的分子轨道”!在一些IF很高的非专业理论化学的期刊上,比如ACS Nano,居然也有作者写“MP2计算的分子轨道能量是xxx”之类明显错误的表述,真不知道审稿人都是什么水平、干什么去了。量子化学里有很多种轨道,不要和分子轨道混淆。比如MCSCF计算完给出的轨道是赝正则分子轨道,Gaussian里对MP2、CCSD等方法加上pop=NO关键词给出的是自然轨道、做轨道定域化处理后得到的是定域化分子轨道(参看http://sobereva.com/380)等等。它们都不是单电子有效势算符的本征函数,不属于分子轨道。本文说的泛函都是指双杂化泛函以外的泛函,双杂化泛函也是得不到分子轨道的,因为它包含类似MP2的一项,因此也脱离了单电子近似。量子化学程序做完双杂化泛函计算后给你的轨道能量其实只是双杂化泛函当中的杂化泛函那部分计算时产生的轨道,并非是双杂化泛函级别的轨道。
5 关于Koopmans定理
前面已经多次提到Koopmans定理,但都没有细说。为了令读者了解得更透,这里专门详细说一下,并且做一些额外的讨论。
Koopmans定理的含义如今经常被广义化,其1934年刚提出来的时候最初的说法是:分子体系的第一电离能等于HF方法的HOMO能量。当然,这段话是忽略了电子相关和轨道弛豫效应情况下的。根据J. Chem. Phys., 150, 074108 (2019)的大量测试,对各种价层电子电离的情况,HF的轨道能量的负值与实验VIP偏离的绝对平均值为1.86 eV,若只考虑最外层电子电离的情况,则误差为0.74 eV。可见至少对于最外层电离,即第一VIP,Koopmans定理能给出定性正确的结果。之所以HF方法精度很烂,但Koopmans定理表现得还不差,这被认为是忽略电子相关和忽略轨道弛豫效应带来的误差往往能很大程度抵消。
Koopmans定理在一些文章和书籍中也被扩展到LUMO的情况,说分子的第一(垂直)电子亲和能等于LUMO轨道能量。但这个近似从实际结果上来看非常糟糕,完全没有实用性。虽然占据分子轨道没有严格的物理意义,但非占据分子轨道的物理意义明显更差,而且能量和形状对基组非常敏感。
Koopmans定理后来被用于DFT领域,为了区分,有时叫DFT-Koopmans定理。这个定理说的是:完全精确的交换-相关泛函做KS-DFT计算得到的HOMO能量等于第一VIP。这个关系可以证明是严格的,而非近似的。此处的第一VIP既可以是指以E(N-1)-E(N)方式算的,也可以是指实验测定的,对于精确的泛函二者显然是相同的(前提是排除了杂七杂八其它因素带来的误差)。
DFT-Koopmans定理对于LUMO并不适用,不能说精确泛函下KS-DFT计算得到的LUMO能量等于VEA。但是在精确泛函下VEA精确等于N+1电子态的HOMO能量的负值。因为-HOMO(N+1)=E(N)-E(N+1),VEA(N)=E(N)-E(N+1),故VEA(N)=-HOMO(N+1)。
DFT-Koopmans定理原本只是用于HOMO,但也有人用它把HOMO以下的轨道与更内层电子电离的VIP相关联起来,确实对于某些泛函这个关系不错,但即便对于精确的交换-相关泛函这也不是严格的。
即便有DFT-Koopmans定理将HOMO能量与实验VIP联系了起来,也绝对不要理解为“实验可以测定分子轨道能量”,仿佛分子轨道能量是真实存在的东西,而应当说成“实验可以测定精确的交换-相关泛函计算出的Kohn-Sham轨道能量”,即要强调实验数据只不过是正好对应于特定情况下计算得出的某种纯理论上定义东西。
由于精确的交换-相关泛函的形式是谁也不知道的,因此我们目前不可能直接基于Koopmans定理靠做个KS-DFT计算就得到严格精确的(第一垂直)电离能以及可能也很准确的更深层电子的VIP。
可能你会看到在一些地方有些人说Koopmans定理只对HF才近似适用、对DFT不适合。还有的人甚至说KS-DFT轨道没有丝毫物理意义,理由是“违背了Koopmans定理”。这些乱七八糟或者严重歪曲的说法要么过时,要么根本没有正确理解KS-DFT或Koopmans定理。实际上,不同的现有的泛函对Koopmans定理满足得有好有坏,这和泛函的HF成份有关。HF成份较高的,以及一些长程校正泛函,其轨道能量的负值比起HF轨道的更接近实验VIP。特别是Bartlett搞的QTP (Quantum Theory Project)为开头的一系列泛函,对Koopmans定理满足得比起现有的几乎其它任何泛函都好(也明显好于HF)。对价层电子电离,QTP17分子轨道能量的负值与实验测定的VIP平均绝对误差为0.5 eV,而换做是其它泛函,即便是表现相对较好(HF成份高达54%)的M06-2X,误差也达到1 eV左右;如果是纯泛函,误差则高达4~5 eV。在计算内核电子的电离能方面,CAM-QTP(00)和QTP17根据Koopmans定理算的甚至比昂贵的IP-EOM-CCSD精度更高。
之前笔者在《使用Multiwfn绘制光电子谱》(http://sobereva.com/478)一文中还提到了广义化Koopmans定理,通过E(N-1)-E(N)方式算的第一VIP来试图消除分子轨道能量与各层电子电离能的偏差,通过此处理,就可以直接用分子轨道能量较好地模拟光电子谱。显然,对于QTP系列泛函来说,这种处理的必要性就没有那么高了(但还是会有改进)。
由于分子轨道本来就不现实存在,所以原理上也没有办法判断轨道能量算得准不准。不过,从实用角度出发,有研究文章通过让占据轨道的能量的负值与电离能尽可能吻合来优化泛函参数。前面说的QTP17和CAM-QTP(00)泛函就是如此。还有其它一些文章,比如J. Comput. Chem., 21, 227 (2000)也是这么考虑,发现把B3LYP的杂化成份从20%提升到60%,就能让轨道能量与实验电离能吻合程度有几倍的改进。通过这样优化的泛函,就可以很省事地计算VIP,即只要在基态极小点结构下做一次单点计算,各层电子的VIP就全都能较好地得到了(这种做法的精度和可靠性,以及与其它各种方法的比较,看J. Chem. Phys. 150, 074108 (2019)中的测试)。泛函的ω参数的调控也很值得一提,这在《优化长程校正泛函w参数的简便工具optDFTw》(http://sobereva.com/346)一文中专门说了。这种做法是通过调节长程校正泛函的ω参数,尽可能令N和N+1电子态同时最大程度满足Koopmans定理,这也是精确的交换-相关泛函应当满足的条件之一。这样的泛函在计算激发能、(超)极化率、fundamental gap、单-三重态激发态能量差等方面比起未经调控的泛函往往明显更好,但缺点是不同体系最优ω参数不同,对每个被计算的体系都要优化一遍ω。
另外,也有文章通过利用分子轨道能量来较好计算VEA。例如J. Phys. Chem. A, 109, 8923 (2005)指出VEA可以近似计算为-E(LUMO)-E(HOMO)-VIP,相当于存在VEA+E(LUMO)≈-VIP-E(HOMO)的关系。至少对于B3LYP而言,通常-E(LUMO)>VEA、VIP>-E(HOMO)。对38个分子,在PBE/aug-cc-pVTZ下用这种方式算的平均绝对误差为0.49 eV。杨伟涛等人对纯泛函提出了一种非经验的global scaling correction (GSC)方法,在Mol. Phys., 116, 927 (2018)里有介绍。将LUMO轨道波函数代入文中14式得到校正量Δε(LUMO),然后VEA就可以计算为-[E(LUMO)+Δε(LUMO)],这比基于纯泛函通过E(N)-E(N+1)的标准方式算的更准确,不过算这个校正量的程序没公开提供。