在Gaussian中计算IRC的方法和常见问题
在Gaussian中计算IRC的方法和常见问题
Methods and FAQs for calculating IRC in Gaussian
文/Sobereva @北京科音
First release: 2018-Jan-4 Last update: 2020-Sep-14
0 前言
IRC是量子化学研究化学反应的重要概念,它是质权坐标下连接势能面相邻两个极小点的能量最低路径,描述了化学过程在不考虑热运动因素下最理想的结构变化轨迹,对于讨论微观化学过程至关重要,而且也是验证过渡态找没找对的最决定性的方法。在无数年前笔者写的《过渡态、反应路径的计算方法及相关问题》(http://sobereva.com/44)中有很多介绍和讨论,建议看看。
在Gaussian中计算IRC的问题是笔者在网上被问及最多的问题类型之一,在计算化学公社论坛(http://bbs.keinsci.com)和思想家公社QQ群里属于半周经问题。为了免得日后反复重复回答大量相似问题,决定写个文章专门说一下。下文先说一下Gaussian中产生IRC的方法,然后再集中回答一下我常被问及的各种与IRC有关的问题。还有很多其它和做IRC有关的知识,限于时间精力以及表达形式的限制就不写在这里了,在笔者讲授的北京科音初级量子化学培训班(http://www.keinsci.com/workshop/KEQC_content.html)里对IRC和相关问题有比本文全面详细得多的讲解,给了十分丰富的例子。在面向已有一定基础的研究者开设的北京科音基础(中级)量子化学培训班(http://www.keinsci.com/workshop/KBQC_content.html)里对这部分内容讲授得明显更深入,并传授更多经验技巧,欢迎参加!
笔者还另外写了一篇和IRC有密切关系的文章《谈谈Gaussian产生downhill路径的功能》(http://sobereva.com/571),是Gaussian的IRC功能的一个特殊用法,推荐在看完本文后阅读。
本文内容适用于Gaussian09和16。
1 在Gaussian中产生IRC的方法
在产生IRC之前,必须先进行过渡态(TS)优化。必须以过渡态的结构为初始结构,才能进行IRC任务。既可以将优化出的TS结构直接写在IRC输入文件里,也可以用geom=check关键词从优化TS任务的.chk文件里读取最后一帧结构。下面是一个最简单、典型的找TS+走IRC的过程:
(a)在gview里凭借化学直觉将体系摆成尽可能接近于过渡态的结构,保存输入文件为TS.gjf。
(b)将TS.gjf里的关键词设为# B3LYP/6-31G* opt(calcfc,noeigen,TS),用Gaussian执行之
(c)打开上一步任务的输出文件,保存成输入文件IRC.gjf
(d)将IRC.gjf里的关键词设为# B3LYP/6-31G* IRC=calcfc,用Gaussian运行即可得到IRC。此任务出现报错几率很大,仔细耐心阅读完本文就知道怎么办了。
IRC任务特别需要注意的一点是,产生IRC和找过渡态用的计算级别必须严格一样!确切来说,任何影响势能面的设定必须严格相同(如果你不知道哪些会产生影响,比较修改前后单点能是否有差异便知)。比如找过渡态用了int=ultrafine scrf(SMD,solvent=ethanol),那么产生IRC的时候也必须写上int=ultrafine scrf(SMD,solvent=ethanol),否则相当于两个任务的势能面并不相同,走IRC也就不是从势能面的准确的一阶鞍点来走的,此时IRC任务要么出错,要么结果无意义。
Gaussian09/16支持多种产生IRC的算法,比较主要的三个在这里说一下:
LQA(Local quadratic approximation):这是非常传统的一种IRC产生算法,1988年提出了,每一步都需要利用Hessian矩阵。
HPC(Hessian-based Predictor-Corrector):2004年由Gaussian作者之一Schlegel提出,这是G09/16默认的算法。这个方法产生IRC每一个点的时候,是将LQA方法作为预测步(predictor),再用修改的Bulrisch-Stoer方法做为校正步(corrector)。这样“预测+校正”结合起来,精度比只用LQA更好,而耗时增加不多。此方法校正步的计算过程是一个迭代过程,众所周知,只要有迭代,就会伴随着不收敛的可能。HPC方法的一个最大问题,正是做校正步的时候经常不收敛,导致网上问Gaussian的IRC问题的多半都是问这个。
GS2:1989年提出,GS是Gonzalez-Schlegel的缩写。这是G03默认的算法,在《过渡态、反应路径的计算方法及相关问题》里有详细介绍。此方法之所以不再是如今版本默认的,是因为此方法耗时比HPC高很多,每一步都相当于做一次限制性优化。
Gaussian里还支持其它方法,诸如DVV、EulerPC等,由于平时用不到,所以就不提了。
走IRC时,对初始结构需通过calcfc关键词来产生精确的Hessian矩阵。LQA和HPC方法走每一步的时候也都需要Hessian矩阵,默认情况下是通过Bofill方法基于梯度和前一步的Hessian近似产生的。如果你在IRC里用calcall关键词代替calcfc,则每一步的Hessian矩阵都会精确生成,显然IRC走得也会更准确,但是耗时会暴增一个数量级甚至更多。也可以在写calcfc的同时用recalc=n关键词,此时每n步才会精确重算一次Hessian矩阵。显然,n越小越准确,耗时也越高,n=1时和calcall是等价的。
走IRC默认是先往反应路径一边走,之后再往另一边走,两边的路径和初始的TS结构合在一起就是完整的IRC。IRC每一帧结构对应一个反应坐标和一个能量,通常将IRC以“能量vs反应坐标”绘制成曲线图表示。在Gaussian中IRC任务的初始结构(过渡态结构)被作为反应坐标的零点,往反应物一侧反应坐标为负,往产物一侧反应坐标为正(但有时也可能是和实际情况反过来的,看后文)。Gaussian给出的反应坐标单位是amu^1/2 Bohr,这里amu是指atomic mass unit,有amu^1/2这项是因为IRC是在质权坐标下定义的。
由于IRC是在质权坐标下定义的,因此结果受同位素质量设定的影响,这里有具体例子展现同位素产生的影响《谈谈温度、压力、同位素设定对量子化学计算结果产生的影响》(http://sobereva.com/423)。如果在IRC里写上Cartesian关键词,代表在笛卡尔坐标下执行IRC任务,此时结果就不叫IRC了,确切来说叫做MEP(minimal energy path,极小能量路径)。笔者时不时看到有的菜鸟莫名其妙地在IRC里写了个cartesian关键词,明明他都不知道这是什么含义就瞎写,明显是被以讹传讹了,而且还是不求甚解就知道盲目模仿。
在IRC里可以用Reverse或Forward关键词让程序只往逆方向或者正方向一侧走IRC。经常有人问诸如怎么我写的是Reverse,但是Gaussian反倒往产物方向走了?或者,我让程序往两边走,走出来的IRC怎么左边是产物右边是反应物?那是因为哪边是反应物哪边是产物只有研究者自己知道,Gaussian知道的只是那是两个不同方向而已。如果你想明确让Gaussian知道指定哪边是正方向哪边是逆方向,需要在IRC里利用Phase关键词定义。
走完IRC后,可以用gview打开输出文件,在窗口左上角切换帧号可观看IRC每一帧结构,或者点绿色圆点播放IRC轨迹。IRC轨迹动画可以通过File - Save Movie - Save Movie File保存出来。IRC的“能量vs反应坐标”图可以用Results - IRC/Path来观看,点击图的左上角的Plots - Plot Molecular Property还可以把几何参数等信息随反应坐标的图绘制出来,在图上点右键还可以将数据点导出,用来在Origin之类程序里绘制和调节达到更好效果。
走IRC有两个参数非常关键:
(1)Maxpoints:设定的是每个方向走的步数,默认为10。比如设Maxpoints=20且让IRC往两边走,则IRC总共最多走1+2*20=41个点,其中1是过渡态那个点。注意maxpoints设的是步数上限,因为走IRC时,如果已经走到了被程序自动判断是离极小点很近的位置了,则IRC就认为这个方向的IRC已经产生完毕了,会提示PES minimum detected,然后就开始走另一头了。如果两头都走完了,就输出汇总信息然后正常结束了。maxpoints设的越大,显然IRC可能走的点数就越多,总耗时也会越高。
(2)Stepsize:设的是IRC步长,默认为10。如果设的是正值,则单位是0.01 Bohr,如果设的是负值,则单位是0.01 amu^1/2 Bohr。比如你设stepsize=20,则步长就是0.2 Bohr,由于默认情况下IRC任务是在质权坐标下做的,因此程序会自动转换为以amu^1/2 Bohr为单位的质权坐标下的步长,仔细看输出文件就能找到相应信息。stepsize设得越小,IRC越准确、曲线越光滑,IRC轨迹震荡情况越不容易出现;stepsize设得越大,则IRC越不准确,曲线越可能出现褶皱,观看IRC轨迹时也越可能看到结构震荡现象,在用HPC方法的时候还越容易出现校正步不收敛而报错的问题。
为了更直观看到stepsize对IRC产生的影响,以及LQA和HPC的差异,这里给出HPC原文里的图,这是对一个模型势能面Muller-Brown surface走IRC的情况:
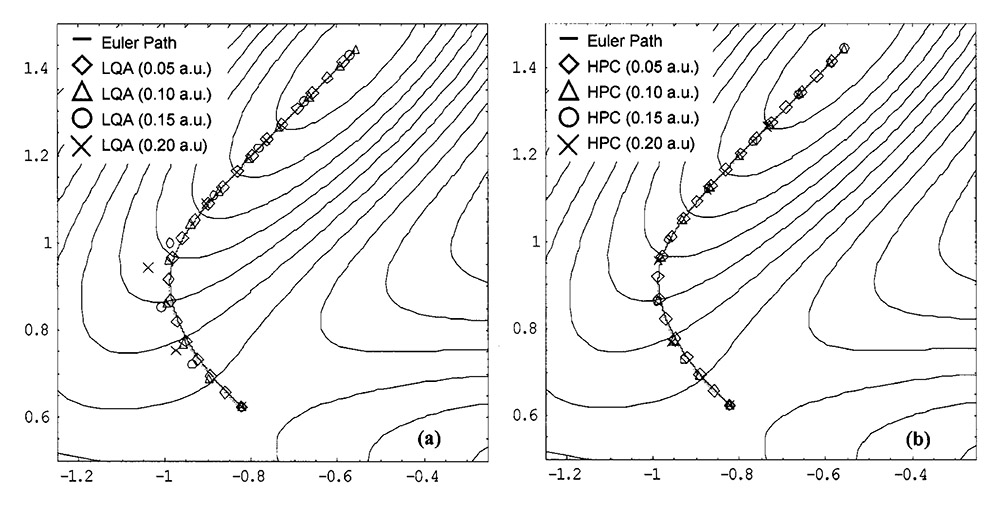
由图可见,相同步长情况下,右图的HPC方法跑出的IRC的点比左图的LQA方法更接近于精确的IRC曲线(实线),因为引入了校正步。而带来的代价就是在实际研究中很容易出现校正步不收敛问题。从上图也看出,IRC步长设得越小,走的IRC点也越接近精确路径,反之误差越大。在LQA方法结合0.2 Bohr步长(默认的stepsize的两倍)时走出的IRC已经误差挺明显了。
maxpoints和stepsize共同决定IRC最多能走多长,即每一侧长度上限是maxpoints*stepsize。默认设定下,IRC只能跑出与TS比较接近的一段,如果你想让IRC跑得更长,显然要么增加maxpoints要么增大stepsize。增大maxpoints会增加耗时,而增大stepsize,则会导致IRC精度下降、HPC方法容易因校正步不收敛而出错。
不同反应的反应路径的总长度是不同的,可能某个设定下对A反应已经把IRC跑得很完整了,但是对于B反应,IRC的两个端点距离反应物和产物极小点还尚有不小距离。要判断IRC是否跑得比较完整,可以看“能量vs反应坐标”两端曲线是否已经接近水平了,也可以看gview在“能量vs反应坐标”图下方给出的“受力vs反应坐标”图,如果两端的点的受力已经比较接近0了,也说明接近极小点了,跑得较完整了。
到底maxpoints和stepsize应该怎么设合适?这要看你跑IRC到底想干嘛,以下是笔者的建议:
(a)验证过渡态找没找对:对于这个目的,IRC只要跑一小段就足够看出趋势了,也不需要IRC跑得质量多高。maxpoints用默认即可,stepsize可以设大到15或20,从而在不增加计算量的前提下能比默认时稍微跑长一些。建议加上LQA关键词避免HPC方法因校正步不收敛而中断。
(b)获得近似的反应物和产物结构:一切同上,但是把maxpoints设50甚至更大,从而使IRC尽可能跑完整。
(c)获得高质量、完整的IRC曲线:这主要是用于发文章目的,粗糙、不光滑的IRC曲线图放到文章中肯定会遭人嫌,我们也希望IRC尽量跑完整从而能够充分描述整个反应历程。为了让IRC质量高,可以把stepsize设小到5。由于设小了步长,又想要IRC完整,maxpoints必须设很大,比如200。如果此时跑出来的IRC还是不平滑,或者因HPC方法的校正步不收敛而中途报错中断,应再加上calcall重新跑。如果计算能力不足用不起calcall,也可以用比如recalc=3或5。
2 一些与IRC相关的常见问题
看这一节之前应确保已经仔细阅读、充分理解了上一节的文字。Q:IRC任务报错啦!末尾提示Maximum number of corrector steps exceded咋办?
A:这就是前面反复说的HPC方法校正步不收敛。有以下办法:
(1)用LQA,比如IRC(calcfc,LQA),由于此时不涉及校正步了,因此100%解决问题。但这容易造成IRC不够准确、不光滑。
(2)如果你不希望用LQA在避免报错的同时牺牲IRC精度,则尝试减小步长(越小避免报错的几率越高),比如用IRC(calcfc,stepsize=5)。或者加上calcall,若嫌太昂贵就改用recalc=x(x越小避免报错的几率越高,但也越昂贵)。
(3)改用GS2算法,即IRC(calcfc,GS2),可完全避免以上报错。耗时比LQA高得多,但精度也比LQA好。
(4)IRC里加上ReCorrect=never,这使得HPC方法不做校正步,故也完全避免以上报错。此时耗时和LQA相同,但所得IRC精度不如LQA,因此强烈不建议用。
(5)IRC里加上maxcyc=N(N应大于默认的20)来加大HPC校正步迭代次数上限。笔者时常在菜鸟的IRC输入文件里看到这关键词,笔者强烈不建议用。虽然此方法不是说解决问题的可能性精确为0%,但可能性实在甚微,没有试的必要。“不收敛就直接加大循环次数上限”是菜鸟最常见的思维方式。
顺带一提,有时笔者看到有人的IRC输入文件里在IRC里用了tight,这是用来把校正步收敛限设得更严的,用这个完全是莫名其妙,也不知道从哪里学来的。本来默认的收敛限下就容易不收敛,居然还给设得更严,明显会导致出现上述报错的几率大增。
Q:怎么IRC刚走了几步就正常结束了?怎么IRC走出来的两侧的曲线是相同的?
A:此问题是继上一个问题在网上被问得最多的与IRC有关的问题。出现这种问题都是因为优化过渡态时定位准确度不够。看下图,当优化出的过渡态位置不准确时,结构就不是在IRC的极大点了,而是稍微偏离一些的红球的地方
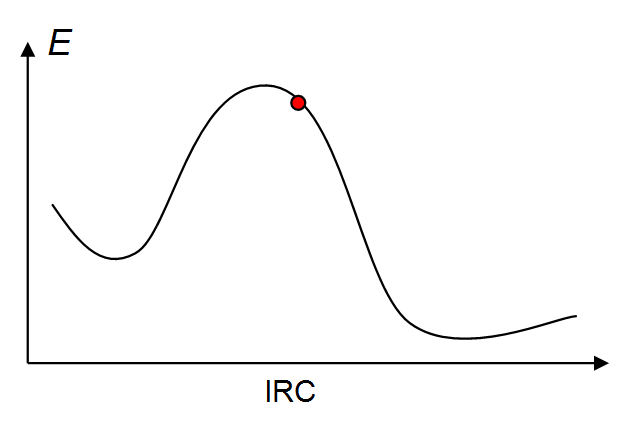
出现这种情况时,往右边产生IRC能正常产生,但是从红球位置往左边产生IRC时,还没怎么走,程序就发现能量升高了,误以为IRC已经走到了离极小点很近的位置,于是就不再继续走了,就正常结束了。还有一种情况,是刚往左边走IRC,由于体系受力是冲着右边的,导致马上转了个弯就往右边走了,就呈现了IRC左右两边曲线都一样的结果。
对这个问题,应按照以下方式排查和尝试解决
(1)先确保初始结构是之前优化TS得到的结构,而且过渡态优化和走IRC都是在严格相同级别下进行的。
(2)提高过渡态定位精度。在找过渡态时候用tight,对于DFT再同时结合int=ultrafine(此时产生IRC也必须用int=ultrafine)。如果还不行,优化过渡态时用calcall(或者用诸如recalc=3)。
(3)如果反复尝试了(2)的方法还是不行,或者你不想尝试(2),毕竟会增加很多耗时,那也可以尝试增大IRC步长,比如20乃至30。由于步长大了,从上图红球的位置往左走的时候可能一下子就越过了TS,之后就能正常继续往左产生IRC了。不过步长大了容易导致HPC校正步不收敛、IRC不准确不平滑等问题,怎么考虑和处理前面已经说了。
另外,出现这种问题还有一种可能是在IRC任务中,基于自动初猜的波函数做SCF后收敛到的波函数与找过渡态任务最终得到的波函数不同,此时相当于IRC任务所在的势能面和过渡态搜索任务所在的势能面不同,这也会导致IRC异常,因为类似于违背了前述的走IRC的“任何影响势能面的设定必须严格相同”的这个前提。出现这种情况时,你会发现IRC任务第一次输出的SCF Done能量和找过渡态最后一步的SCF Done能量明显不同。为解决此问题,走IRC的时候可以用guess=read关键词,从优化过渡态的chk文件中读取最后的波函数(并且最好用forward和reverse关键词通过两个任务分别跑正向和逆向IRC),这样通常可以确保IRC任务所在的势能面和优化过渡态时相同。
用SMD溶剂模型时,也可能个别时候由于数值噪音问题出现IRC走几步就停了的现象。可将优化和走IRC用的溶剂模型都改为IEFPCM再试,说不定能解决。
Q:我的IRC怎么成这样了?怎么有个点跑到下头去了?

A:即便IRC任务还在算着,也可以用gview打开输出文件看看走到第多少步了,检查是否正常。当IRC还没有走完的时候就打开输出文件,或者看的是IRC失败的任务的输出文件,往往就会看到中间有一个点突然掉下去的现象。任务正常结束后再看就不会有这个现象。
Q:走出来的IRC最两端的点是反应物和产物结构么?
A:否。IRC和几何优化不一样,几何优化的步长是随机应变的,而IRC的步长是固定的。从TS开始走,即便把maxpoints设得非常大,stepsize设得很小,来让IRC跑得又完整质量又高,也注定不可能恰好有一个IRC点正好落在与反应物或产物对应的极小点的位置。因此,必须对IRC两端的点进一步做几何优化才能得到准确的反应物或产物结构。
Q:为什么我看IRC趋势是对的,我对IRC两边的点优化,优化得到的结构却不是我想要的极小点结构?
A:有两种可能:
(1)与这个TS直接连通的极小点本来就是那样。由于你的IRC跑得还太短,在加上你的化学直觉不准,导致并没有估计对真正极小点结构。可以让IRC尽量跑完整一些来检验。
(2)优化的时候由于走得步子太大,或者Hessian矩阵不准确,导致优化到了其它邻近的极小点去。此时可以在优化时用较小的步长上限并结合notrust(相关信息看《量子化学计算中帮助几何优化收敛的常用方法》http://sobereva.com/164),或者结合calcall/recalc=x提供精确Hessian矩阵,或者先把IRC跑得尽量长一些,使两端结构尽可能接近极小点时再做几何优化。
Q:柔性扫描和IRC有什么区别?柔性扫描能代替IRC么?
A:二者概念完全不同,不能代替。IRC是从TS开始顺着虚频方向,沿着梯度负方向移动结构走出来的轨迹,所有坐标是同步变化的。而柔性扫描,是令某个(或某些)坐标以特定方式不断改变,每次都优化其余所有变量所得到的(限制性优化),故所有变量间的变化不是协同的,得到的能量变化曲线与IRC也是大相径庭的。柔性扫描曲线及其轨迹经常会出现突跃,而不像IRC那样总是平滑变化。不过,当某个化学过程主要只涉及一个变量发生变化的话,柔性扫描这个变量和走IRC产生的曲线倒是比较相近,此时如果你用的计算级别没有解析Hessian而不容易走IRC的话,用柔性扫描也未尝不可(比如当年没有TDDFT解析Hessian的时代,由于不方便找TS走IRC,对激发态质子转移过程的研究经常是靠柔性扫描来代替)。
关于柔性扫描,建议阅读《详谈使用Gaussian做势能面扫描》(http://sobereva.com/474)。
Q:我的IRC曲线在过渡态位置怎么特别尖/不平滑,貌似不对劲?
A:应当确认初始结构是优化过的过渡态结构,并且找过渡态和走IRC用的计算级别(包括任何能够影响单点能的因素)严格相同。如果你就是这样做的,那么尝试增加过渡态的定位精度,比如优化过渡态时用tight收敛限、calcall/recalc=x、增加积分格点精度等。如果这些也都试了,走IRC时用GS2算法,或加上calcall试试。另外也要确保IRC任务第一步收敛到的波函数与过渡态搜索最后一步相同,这在前面已经说过了。
Q:IRC任务如何续算?
在IRC里添加restart关键词即可,例如IRC(restart,maxpoint=15) b3lyp/6-31g(d)。此时%chk应当和之前中断的IRC任务相同。
Q:我发现之前IRC跑得不够长,如何在不完全重算的前提下在两侧再增加一些点?
A:比如maxpoints=15的设定下IRC任务已经正常跑完,现在还想两个方向各增加5个,执行IRC(restart,maxpoint=20),%chk应当和之前的IRC任务相同。
注意gview观看restart得到的IRC轨迹要打开chk/fch文件,而不要打开out/log文件,否则看到的只是续跑出来的点。
Q:IRC任务出现L502错误怎么办?
A:这是走IRC过程中出现了SCF不收敛所导致。按照常规解决SCF不收敛的方法尝试添加适当的关键词解决,见《解决SCF不收敛问题的方法》(http://sobereva.com/61)。除此以外,如果是IRC任务第一次做SCF就没收敛,而且之前opt任务的chk文件还在,可以做IRC任务时加上guess=read读取其中收敛的波函数当初猜。如果是IRC中途某个点SCF不收敛,也可以减小IRC步长,因为IRC的每个点会自动沿用上个点收敛的波函数当初猜,显然二者结构相差越小,那么上个点收敛的波函数就是这个点越好的初猜波函数。
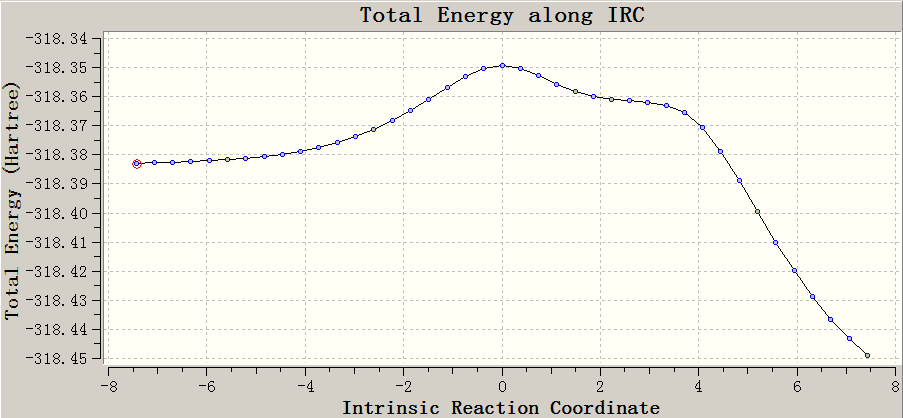
Q:我的IRC看起来比较怪,合理么?怎么边上还凸起来一块?(下图为笔者随便找的例子)
Q:我的理论方法支持解析梯度但不支持解析Hessian(比如CCSD),怎么跑找TS并且跑IRC?
A:先说此时怎么找TS。有两类做法:
(1)纯依赖于梯度的方法
用CCSD/cc-pVDZ opt(TS,modRedundant,noeigen)或者opt(TS,gediis,noeigen)或QST2/3,这些过渡态搜索关键词都不需要提供初始Hessian
(2)基于半数值Hessian的方法
把初猜TS结构的Hessian矩阵存到chk文件里:# CCSD/cc-pVDZ freq(这步耗时很高)
从chk里读Hessian矩阵并优化TS:# CCSD/cc-pVDZ opt(TS,noeigen,readfc)
找到TS结构后,使用这个关键词即可生成IRC:CCSD/cc-pVDZ IRC(gradientonly,euler)。这里gradientonly代表使用只依赖于梯度的IRC算法,默认是DVV方法,由于结果烂到爆,所以改成相对好点的Euler方法,但质量也不怎么样,经常震荡得很厉害。
Q:我怎么对IRC过程做波函数分析,考察电子结构随反应过程的变化?
A:参看《产生Gaussian的IRC和SCAN任务每个点的波函数文件的工具》(http://sobereva.com/199)、《通过键级曲线和ELF/LOL/RDG等值面动画研究化学反应过程》(http://sobereva.com/200)。
Q:我之前对过渡态已经做过振动分析了,chk里已经有了过渡态结构下的Hessian矩阵,能否在IRC计算时直接读取之,避免用calcfc计算Hessian以节约时间?
A:完全可以,对较大体系也推荐这么做。在IRC里加上rcfc关键词即可,此时就不需要写calcfc了,程序会从%chk文件中直接读取Hessian矩阵来用。