由Mulliken布居和Lowdin布居再谈旋转不变性问题
由Mulliken布居和Lowdin布居再谈旋转不变性问题
Talking again rotation invariance of Mulliken population and Lowdin population
文/Sobereva @北京科音 2010-Feb-20
Talking again rotation invariance of Mulliken population and Lowdin population
文/Sobereva @北京科音 2010-Feb-20
前言:
最近看到两篇Mayer的讨论Lowdin布居旋转不变性问题的文章Int. J. Quant. Chem, 106, 2065-2072和CPL, 393, 209-212,受到启发,继之前写的《谈谈原子轨道朝向引起的量化计算结果与经验观念的差异》(http://sobereva.com/52),本文对旋转坐标问题作进一步讨论。注:本文说轨道时是指原子轨道,极小基与之对应。说原子内基函数时,则含义更为普遍,包括用了扩展基的情况,此时与原子轨道无对应关系。
1. 基本概念回顾
基函数的变换可以分为线性和非线性,常见的都是线性变换,此时变换关系可以用变换矩阵X来表示。设有a基函数集a(1),a(2)...和b基函数集b(1),b(2)...变换矩阵为X;共轭矩阵为X`。则有b(i)=∑[k]X(k,i)a(k)。若有一个算符Y,a为基时叫O,即O(i,j)=<a(i)|Y|a(j)>;b为基时叫P,即P(i,j)=<b(i)|Y|b(j)>。则有P=X`OX。若有一个波函数在a为基时以向量c表示,在b为基时以c"表示,则有c=Xc"。基函数的酉变换是线性变换的特例,只有当变换前后都是正交归一集时线性变换才是酉变换,此时X是酉矩阵,即XX`=I。由∑[k]|a(k)><a(k)|=1和∑[k]|b(k)><b(k)|=1的关系可推得矩阵元X(i,j)=<a(i)|b(j)>。
基函数的线性变换在量子化学计算中经常用到,例如由原来不同类型轨道变换为杂化轨道(如s、p轨道组合成sp3轨道),由扩展基变换为自然原子轨道或AOIM轨道,由非正交基函数变换为正交基,由原子基函数变换为群轨道成为分子所属点群不可约表示的基等等。本文涉及的是由分子旋转(即朝向改变)导致的基函数变换,这里只讨论笛卡尔型高斯函数为基函数的情况,如果是STO、球谐高斯函数,则实际上分子旋转引起的是其角度部分波函数原点的变化。
有的量化方法有基函数线性变换不变性,例如《谈谈原子轨道朝向引起的量化计算结果与经验观念的差异》已经证明了HF方法的这个特点,在推导中只利用了线性变换的规则。而有的量化方法不具备基函数线性变换不变性,只具备酉变换不变性,Lowdin布居就是如此。
一些引起基函数线性变换的操作在某些条件下可回归为酉变换,尤其是分子的旋转操作这点很重要。了解了这些条件,才能正确地使用量化方法。下面先谈谈重要的原子基函数正交性问题。
2. 原子基函数的正交性
类氢原子轨道:原子内轨道都正交。STO(Slater type orbitals):相对于类氢原子轨道修改了径向部分的形式,使其波节消失,每个STO仍对应于一个原子轨道(虽然高精度计算时也可以用多个STO展开一个原子轨道)。不同类型(指s、p、d...后同)之间的轨道都正交;相同类型不同主量子数轨道间不都正交,不正交情况例如3px与4px。
球谐型GTF(Gaussian type function):将STO的径向形式修改为了r^n*exp(-α*r^2),与原子轨道主量子数不再相关,紧缩或弥散只取决于α值,使用扩展基时与原子轨道无一一对应关系,一般用壳层来描述,一个壳层是指结合一个特定的轨道指数的类型相同的几个基函数,在此不考虑SP壳层这样的类型混合的特例。相同壳层内的基函数都正交。不同类型的不同壳层间的基函数都正交,相同类型的不同壳层见基函数不都正交,不正交情况例如分裂价基的两套壳层间的角度部分行为相同的轨道。
笛卡尔型GTF:将球谐型GTF的角度部分改为x^i*y^j*z^k形式。与球谐型GTF正交性的区别是对于d、f、g...类型的相同壳层内的基函数不都正交,例如d型的XX、YY、ZZ间不正交、f型的XXY与YZZ不正交等等。这是因为对这些情况,将重叠积分中两个笛卡尔型GTF相乘化为一个GTF后,其i、j、k项都是偶数,所以x、y、z方向积分分量都不为0,故相乘也不为0。球谐型GTF可由笛卡尔型GTF组合得到,详见《谈谈5d,6d型d壳层波函数与它们在高斯中的标识》(http://sobereva.com/51)和Int.J.Quant.Chem,54,83-87。
在分子中,以上这些原子基函数由于原子间的重叠,导致原子间的基函数不正交。但注意对于一些半经验方法,如AM1、PM3,用了重叠矩阵元S(i,j)=δ(i,j)的近似。
3. 旋转分子引起的基函数的变换
上一篇文章已提到,令坐标轴不动而旋转分子,等于令分子不动而旋转坐标轴,由于GTF与坐标轴是绑定的,所以等于对基函数进行了线性变换。变换过程并不会令原子间的基函数发生混合,也不会令不同壳层间的基函数发生混合。如果将基函数按顺序排列,则变换矩阵是对角块形。例如6-31G*(笛卡尔型GTF)的H2O分子因旋转引起的基函数线性变换矩阵X: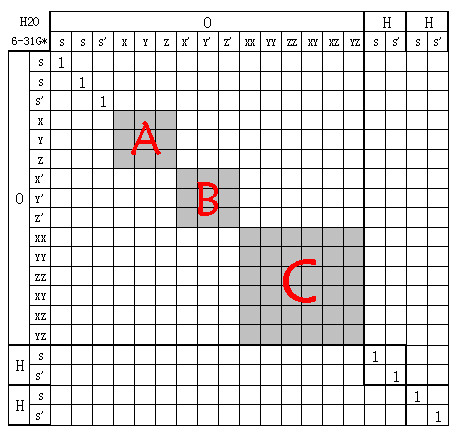
图中'符号用于区分分裂价基的两个壳层。灰色区域是未知的不一定是0的值,是多少以及是不是0取决于怎么旋转。图中基函数的排序是为了紧凑,描述方便,当然实际量化程序中的基函数排序并不一定是这样,但并不会对下面的讨论结果有什么影响。
现在讨论这个矩阵什么时候是酉矩阵。由于旋转不会改变基函数之间的正交性,如果之前全部原子基函数都正交,旋转后显然也都正交,毫无疑问这必然是酉矩阵。但这个条件很难满足,实际上,旋转操作的变换矩阵是酉矩阵的条件没有这么强硬。从图中可见,由于旋转只引起壳层内轨道的混合,而壳层间的矩阵元为0,所以只要每个壳层内基函数都正交,则整个变换矩阵就是酉矩阵。同原子的壳层间基函数不完全正交,以及不同原子间基函数不正交这都没有关系,在旋转操作中不会“暴露”出来。
可以容易地做出证明。设图中三个灰色子矩阵分别叫A、B、C,分别对应于三个壳层内基函数的变换。假设使用的基组满足壳层内轨道正交时,则A、B、C矩阵都是酉矩阵。根据分块矩阵相乘规则,XX`的三个对角块就分别是AA`、BB`、CC`,显然都为单位矩阵,故XX`也为单位矩阵,即X为酉矩阵。
前面提到了,球谐GTF在壳层内基函数正交,所以用这样基函数时X为酉矩阵,用STO、类氢原子轨道时也是。而用笛卡尔型GTF,如果涉及到d及以上角动量壳层时由于壳层内基函数不完全正交,则旋转引起的只能说是线性变换。例如上图中用的是6d轨道,壳层内不都正交,故CC`!=I,所以此时XX`!=I,若在Gaussian中加上5d关键词,即使用球谐型d轨道,则图中的变换矩阵就是酉矩阵了。
4. Mulliken布居、Lowdin布居与的旋转不变性问题
Mulliken布居可以写成矩阵形式,P为密度矩阵,S为重叠矩阵,则PS矩阵的第(i,i)矩阵元就是第i个基函数的Gross原子布居数,也就是这个基函数被Mulliken布居分配到的电子数,显然∑[k∈α](PS)(k,k)就是α原子的Mulliken电荷。Lowdin布居就是做Mulliken布居之前先用Lowdin正交化方法使全部基函数之间正交化,变换矩阵为X=S^(-0.5)。设P'、C'、S'为正交化后基函数的密度矩阵、系数矩阵(只含占据轨道的展开系数,矩阵元i,j为第j个分子轨道第i个基函数系数)和重叠矩阵,则P'S'=P'=C'C'^(T)=X^(-1)CC^(T)X^(-1)^T=S^(0.5)CC^(T)S^(0.5)^T=S^(0.5)PS^(0.5),所以S^(0.5)PS^(0.5)的第(i,i)个矩阵元就是Lowdin布居给基函数i分配的电荷。注意由于AM1、PM3等半经验方法对重叠矩阵近似为单位矩阵,此时Lowdin布居与Mulliken布居是等价的。电荷布局方法应当具备旋转不变性,否则分子一旋转,原子电荷就变了,尤其还会出现等价原子电荷不相等的情况,这是完全没有物理意义的。Mulliken布居得到的原子电荷具备旋转旋转不变性,只需证明旋转后的∑[k∈α](P'S')(k,k)与旋转前的∑[k∈α](PS)(k,k)相等即可。
后面的证明需要用到如下关系:
S'=X`SX。当X为酉矩阵时,S'^(0.5)=X`S^(0.5)X,证明见CPL,393,p209-212。
P'=X^(-1)PX`^(-1)。当X为酉矩阵时,可进一步写为X`PX。
Mulliken布居所得α原子电荷Q(α)=∑[k∈α](PS)(k,k)=∑[k∈α](X^(-1)PX`^(-1)X`SX)(k,k)=∑[k∈α](X^(-1)PSX)(k,k)。
接下来要利用X矩阵为原子对角块矩阵的性质进行变换。上式继续写为∑[k∈α]∑[m]X^(-1)(k,m)(PSX)(m,k)=∑[k∈α]∑[m]∑[n]X^(-1)(k,m)(PS)(m,n)X(n,k)。由于只有m∈α且n∈α时式中X^(-1)(k,m)、X(n,k)才不为0,所以改写为∑[k∈α]∑[m∈α]∑[n∈α]X(n,k)X^(-1)(k,m)(PS)(m,n),这就是求X、X^(-1)、PS三个矩阵的原子α的对角块相乘的矩阵的迹,由于X和X^(-1)的原子α的对角块相乘为单位矩阵而被消掉,所以最终得到∑[n∈α](PS)(n,n),可见与旋转前的∑[k∈α](PS)(k,k)是等价的。这就证明了Mulliken布居得到的原子电荷具备旋转不变性。
上面的证明只用了X矩阵为原子对角块的特点,而X不仅有这个特点,由于壳层间不会混合,故更进一步有壳层对角块的特点,所以可以完全相同地证明Mulliken布居分配给每个壳层的电荷不随旋转而改变。但每个壳层内,比如P壳层的X、Y、Z轨道之间经旋转是混合的,所以分给它们的电荷会经旋转而改变。但如果是绕比如Z轴旋转,则Z轨道不参与混合,X矩阵中3*3的P壳层的块就又分成了2*2的XY的块和1*1的Z的块,通过同样方法又可证明,旋转后P壳层内只有X、Y轨道上的电子数改变,但二者的总和不变;Z的电子数不变。
再来考察Lowdin布居,先假设X为酉矩阵的情况,看看是否经旋转后原子电荷也不变,即能否令∑[k∈α](S'^(0.5)P'S'^(0.5))(k,k)回归到∑[k∈α](S^(0.5)PS^(0.5))(k,k)。
Q(α)=∑[k∈α](S'^(0.5)P'S'^(0.5))(k,k)=∑[k∈α](X`S^(0.5)XX`PXX`S^(0.5)X)(k,k)=∑[k∈α](X`S^(0.5)PS^(0.5)X)(k,k),然后如同证明Mulliken布居的过程一样,利用X可视为原子对角块矩阵特点,可写为∑[k∈α](S^(0.5)PS^(0.5))(k,k)。所以,旋转不会令Lowdin布居所得原子电荷改变。
当X不为酉矩阵时,由于S'^(0.5)!=X`S^(0.5)X,上面的推导第一步就不过去。所以只有使用球谐型GTF、STO来满足旋转操作的变换矩阵为酉变换条件时Lowdin布居才有旋转不变性,而用了6d、10f这样笛卡尔型GTF基函数时则原子电荷会受分子朝向影响。由于X不仅是原子对角块更是壳层对角块矩阵,故Lowdin布居是否具有壳层上电子数的旋转不变性也依赖这个条件。
以上的结论是容易在Gaussian中自行验证的。Mulliken布居是默认输出的,用iop(6/80=1)可以输出Lowdin布居。使用含d基函数的基组,随便计算一个分子(朝向不要摆正),对比加nosymm与不加nosymm(会令朝向不同)搭配5d和6d(若含f则也考虑7f与10f)的情况,就可以验证上述结论。但可能由于数值精度和舍入误差问题使结果末位有一点点差异,不应把这种情况作为不符合旋转不变性的例子。Lowdin布居尽管在5d下不符合旋转不变性,但旋转并不会令电荷变化太大,一般仅在0.01数量级以下,也可能正是因为改变的微不足道而使大家长期忽视了这个问题。而随着原子的6d、10f型轨道更多的加入基组,这个问题会变得相对更明显一些。实际上Lowdin布居提出的年代用的还多是STO基组,并没有这个问题,只是后来大家将之沿用到笛卡尔型GTF基组上,却忘了考虑这个问题。
5. 基函数变换操作对Mulliken布居和Lowdin布居的一般性影响
可以将X对Mulliken布居和Lowdin布居的影响一般化考虑:X是什么样对角块矩阵决定了哪些基函数会混合,变换操作一定会使块内每个基函数的电子数发生变化;保持块内基函数电子数之和不改变,对Lowdin布居X必须是酉矩阵,而对Mulliken布居X没这个要求。X的形状影响X成为酉矩阵所用基函数条件,仅当每个块内基函数都正交时X才为酉矩阵。由此可以将问题拓展考虑。假设旋转会使同原子内的壳层混合,即令X不具备壳层对角块特征,则想让X为酉矩阵从而不令旋转改变Lowdin布居的原子电荷,则不得不使用在原子内完全正交的基函数;而Mulliken布居则可以随意选用原子基函数种类。还可以进一步考虑其它基函数变换操作而不限于分子的旋转。例如要将极小基的CO2分子中每个原子的2px和2py和2s轨道组成不正交的杂化轨道(这里不以常见的sp2方式杂化,因为三个sp2轨道间正交),则变换矩阵X对角线上除了1以外有三个3*3的块,所以这三个轨道杂化前后电子数一定会变。尽管每个原子的2px、2py、2s之间正交,但变换后的三个杂化轨道间不正交,所以小块不是酉矩阵,故而X不是酉矩阵,所以用Lowdin布居时三个杂化轨道的布居数之和不等于2px、2py和2s轨道的和。而Mulliken布居的不变性对X是不是酉矩阵没要求,所以那三个轨道杂化前后布居数总和不变。
旋转不变性是个容易忽视但又重要的问题,在开发新的量化方法时,应当检验方法是否具备这个性质。在实际应用中如果发现某些方法存在这个问题,则不妨撰文讨论,以引起更多人的重视。