详谈Multiwfn支持的输入文件类型、产生方法以及相互转换
详谈Multiwfn支持的输入文件类型、产生方法以及相互转换
Detailed introduction of Multiwfn input files, methods of production and their interconversion
文/Sobereva @北京科音
First release: 2017-May-31 Last update: 2024-Sep-10
0 前言
虽然在Multiwfn入门贴《Multiwfn入门tips》(http://sobereva.com/167)、Multiwfn手册2.5节中已经讲过应该怎么选择合适的Multiwfn输入文件,但还是有一些初级量子化学研究者搞不明白,由于使用错误的输入文件导致Multiwfn自动退出或得不到想要的结果。另外经常有人问如何产生Multiwfn要用的wfn文件之类问题,虽然在Multiwfn手册第四章开头已经明确说明了,但肯定也被他们无视了。此文就把怎么选择合适的输入文件,以及怎么产生它们完整详细地讲一遍。虽然此文主要是给初级用户看的,但有一定经验的用户也建议阅读一下,有些细节可能之前自己疏漏了。另外,本文也顺带把Multiwfn的转换文件格式功能介绍一下,这非常有用。
本文内容对应Multiwfn最新版本,有些内容不适合之前的版本。Multiwfn最新版本可在其官网http://sobereva.com/multiwfn免费下载。不了解此程序者建议阅读《Multiwfn FAQ》(http://sobereva.com/452)。
1 Multiwfn的各种功能所需要的信息
Multiwfn的不同功能需要的信息不同,而不同类型的输入文件能提供的信息也不同,因此不同功能需要不同的输入文件,没法一概而论。由于Multiwfn的功能极多,灵活度极高,所以这里没法把每个功能可以用的输入文件类型一一直接列出,而只能告诉读者选用规则。
Multiwfn的绝大多数功能所需要的信息可以归为四大类:
(1)原子坐标:虽然Multiwfn是波函数分析程序,主要对象是波函数,但也有很多分析只需要有原子坐标就够了,比如做基于promolecular的RDG分析考察弱相互作用(http://sobereva.com/68)、aRDG分析(http://sobereva.com/186)、计算原子配位数(手册3.100.9节)、计算HOMA/Bird芳香性指标(http://sobereva.com/176)等等。
(2)GTF信息:这里说的GTF具体是指primitive Gaussian type function。“GTF信息”具体是指各个轨道的占据数与能量、各轨道向各个GTF的展开系数,以及各个GTF的具体定义(指数、所属中心、类型等),这需要有一定量化理论基础知识才能彻底弄明白。Multiwfn中直接基于实空间函数(即电子密度、静电势、ELF、RDG、自旋密度等各种三维函数,以及相关穴、源函数等六维函数)的分析都是基于GTF信息做的。因此,比如绘制各种实空间函数的曲线图、平面图、等值面图及差值图,做拓扑分析,做盆分析,计算拉普拉斯键级与模糊键级,计算ADCH/CHELPG/Hirshfeld等电荷,做定量分子表面分析,进行模糊原子空间分析等,都需要提供GTF信息。
(3)基函数信息:具体是指各个轨道的占据数与能量,各个轨道向各个基函数的展开系数,以及基函数的具体定义(所属中心、收缩度、收缩系数、指数等)。Multiwfn中大量分析都是基于基函数信息做的,这些分析基本都与实空间函数无关,比如计算Mayer/Lowdin/Mulliken/多中心键级,绘制PDOS/OPDOS,计算轨道成份,做CDA分析,做Pipek-Mezey轨道定域化等。
(4)格点数据:格点数据就是指某个实空间函数在某个三维空间区域中均匀分布的各个格点上的值。一些Multiwfn的功能需要这些数据,最典型的就是主功能13,是格点数据处理功能,可以对格点数据进行运算、统计、提取、按照特定规则屏蔽掉某些区域等操作。格点数据可以从外部文件中读入,也可以通过Multiwfn的主功能5等功能计算得到。
Multiwfn还有大量功能,所需要的信息不属于上述情况,情况比较杂。比如绘制光谱图时,需要各个跃迁的能量、强度等信息,可以从Gaussian输出文件等来源中读取。做AdNDP分析或者NAO方式做轨道成份分析时,需要读取Gaussian内嵌的NBO模块或独立的NBO程序的输出信息。做ICSS计算时需要从Gaussian的NMR任务的输出文件中读取各个位置磁屏蔽张量。
有些功能输入文件有多种选择。比如做CDA分析时,轨道系数可以从Gaussian输出文件中读,也可以从提供了基函数信息输入文件中读。有的分析需要提供多种输入文件,从中读取所需的不同信息,比如做NTO或空穴-电子等分析时,需要从Gaussian输出文件等来源中读取激发态的组态系数,且需要从另外的文件中读取基函数信息。
2 输入文件的选择
不同功能需要不同信息,怎么选择合适的输入文件使得Multiwfn能正常分析?在Multiwfn手册2.5节有个表,这里摘录过来:
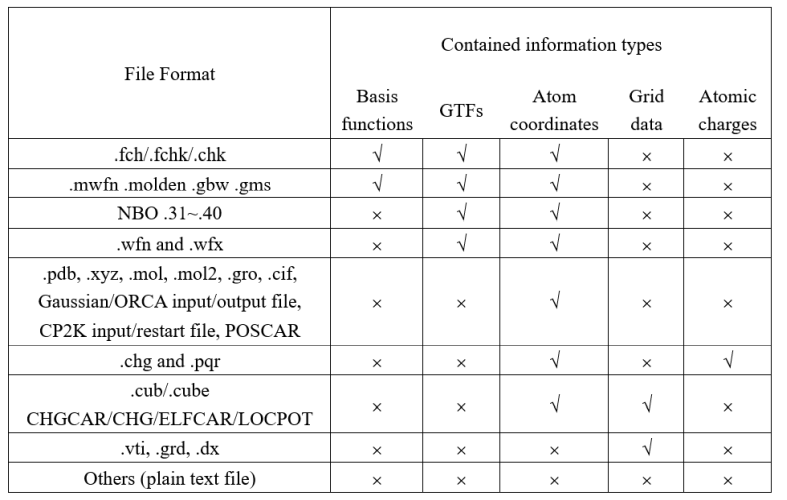
(这一段特别重要,一个字一个字认真看!)表中可看到各种Multiwfn支持的输入文件包含的信息,对勾代表包含,叉代表不包含。比如,我们看到,wfn和fch文件都包含GTF信息,但只有后者包含基函数信息,因此,对于那些只需要GTF信息的功能,用wfn和fch文件都行(显然mwfn、molden等也都可以),结果是一样的;而对于需要基函数信息才能做的分析,则不能用wfn文件。要明白,基函数信息属于比GTF信息更高一级别的信息,含有基函数信息的文件在Multiwfn载入时会自动转化出GTF信息,因此表中凡是能提供基函数信息的文件也都能提供GTF信息。所以,但凡Multiwfn博文、手册里用wfn文件的地方,显然都可以用fch、mwfn、molden等文件代替(当然,前提是你用的其它格式的波函数文件里存的轨道和例子里用的wfn文件里存的轨道是相同的)。再顺带强调一下:倘若你会,甚至已经得到了fch文件,自然就没必要非得刻意效仿博文/手册中的一些例子用out=wfn关键词产生wfn文件(更何况就算你确实出于特殊目的想得到wfn文件,用Multiwfn载入fch文件后也可以用主功能100的子功能2导出成wfn文件)。
Multiwfn的手册十分详细、贴心,在手册第三章介绍每个功能的相应小节的末尾,一般都注明了此功能需要的信息。手册3.x节标题末尾的括号里的数字对应于主功能编号,如果有三级标题,那么标题末尾的数字对应于相应主功能中的子功能编号。比如拉普拉斯键级是Multiwfn当中主功能9的子功能8,因此介绍此功能的小节是3.11 Bond order analysis (9)中的3.11.7 Laplacian bond order (8)。在这一节末尾我们可以看到Information needed: GTFs, atom coordinates,因此,计算拉普拉斯键级用上面图中GTFs那一列下面打着对勾的文件类型都可以,即fch、molden、wfn等。
还有些功能介绍的小节的末尾没有明确注明Information needed,这是因为在这一段落的文字中,已经十分明确写明了应该提供什么文件作为输入。通常这种情况所需要的信息没法简单归结为本文第1节列的那四种。比如通过完全态求和(SOS)方法计算(超)极化率的那一节(3.200.8)中,已经明确有"Input file"这么一段,说明了必须用Gaussian的激发态计算的输出文件,或者记录了跃迁偶极矩和激发能信息的文本文件作为输入。
如果你超懒,或者对英语有心理恐惧,因而死活不愿意/没有能力哪怕稍微看一下手册第三章,那最最起码也得看一下相关博文(在前述的《Multiwfn入门tips》里有汇总)或者手册第四章对应的例子,看看例子里用了什么输入文件,用相同的肯定不会有问题。当然,例子里显然不可能把所有可以用的输入文件全都列举一遍,要不得啰嗦死。经常看到有人比如试图用fch文件绘制光谱图,然后说什么程序闪退,着实匪夷所思!手册、博文里从来没提过可以这样做!
上面图中还有个plain text file,这是指文本文件,即可以用文本编辑器打开能被人来阅读的文件。这没有具体指代,像量化程序输出文件就属于此类,能提供的信息视具体文件而定,反正肯定没法提供基函数/GTF/原子坐标/格点数据。虽说Gaussian输出文件中也有原子坐标,用gfinput关键词还能输出基函数定义等等,但是Multiwfn根本不会去读它。因此,拿Gaussian输出文件作为输入文件,想绘制电子密度图、计算键级什么的,完全是不可能的。
注:如果将settings.ini里的iloadGaugeom设为1,则Multiwfn会从中读取最后一次输出的几何结构(有输入朝向的优先读输入朝向的,没有的话就读标准朝向的),因此之后可以做只依赖于原子坐标的任务。
3 Multiwfn支持的输入文件类型及产生的方法
本节把Multiwfn支持的能提供原子坐标/GTF信息/基函数信息/格点数据的输入文件的基本特征介绍一下,并介绍对于常见情况怎么产生相应的输出文件。
Multiwfn是按照文件后缀名来判断按照什么格式来载入的。对于记录含有GTF信息/基函数信息的文件(统称为波函数文件),Multiwfn目前最高支持到h角动量(说浅显一些,对于比如碳原子,能支持到高至cc-pV5Z基组下产生的波函数),并且只支持Gaussian函数(不支持ADF等程序用的STO函数)。
3.0 mwfn文件
这是笔者提出的目前最理想的记录电子波函数的文件格式,包含了几乎所有波函数分析所需要的信息,并且具有简约、紧凑、可扩展的优点,后文提到的其它格式存在的各种问题在mwfn格式中都没有。mwfn文件从Multiwfn 3.7版开始支持载入和导出。此格式定义的思想、格式的详细说明以及与其它同类文件的对比见笔者发表的此文https://doi.org/10.26434/chemrxiv.11872524。由于mwfn格式相对于其它同类格式具有的显著优点,在未来必会得到越来越多的量子化学程序的支持。
3.1 wfn文件
wfn文件格式最早源于AIM分析程序AIMPAC,现在已经是被支持最广泛的记录GTF信息的波函数文件格式,Gaussian、ORCA、GAMESS-US、Q-Chem、NWChem等诸多程序都可以直接产生它。此格式的详细介绍看此文《高斯fch文件与wfn波函数文件的介绍及转换方法》(http://sobereva.com/55)。注意此文件不包含空轨道,所以没法基于wfn文件考察空轨道,也没法基于wfn文件算一些涉及到空轨道的量,比如计算局部电子亲和能。下面介绍几种常用的量化程序产生wfn文件的方法。
(1)用Gaussian产生wfn文件
在Gaussian中产生wfn文件很简单,写上out=wfn,然后末尾空一行写上输出路径就完了,比如下面是Linux下输入文件一例,在各种计算都完成后,wfn文件就会导出到/sob/poi.wfn
# B3LYP/6-31g(d) out=wfn
[空行]
Title Card Required
[空行]
0 1
O 0.00000000 0.00000000 0.11472000
H 0.00000000 0.75403100 -0.45888100
H 0.00000000 -0.75403100 -0.45888100
[空行]
/sob/poi.wfn
[空行]
[空行]
对于目前的Gaussian版本,后HF或CIS/TDDFT电子激发计算时默认是产生自然轨道并把它们输出到wfn文件里的。如果你用的是MCSCF方法,还必须同时写pop=no,此时得到的wfn文件里记录的才是相应的自然轨道,否则记录的是赝正则轨道。
如果你用的是G09 C.01以前的版本,注意阅读以下文字:
对于后HF计算,如果你想让wfn文件里存的是后HF级别的自然轨道,而不是HF轨道;或者对于CIS/TDDFT计算,想让wfn文件里存的是激发态的自然轨道,而不是基态轨道,那么必须额外写上density关键词(等价于density=current),代表产生当前计算级别的波函数(以自然轨道方式描述),例如# CCSD/cc-pVTZ out=wfn density会在wfn文件里写入CCSD的自然轨道。对于G09 C.01及以后版本,就不再需要写density关键词了,因为程序只要看到out=wfn,就默认用了density关键词。
如果是基于非限制性HF波函数做的后HF计算,你想输出到wfn中的是自然自旋轨道(占据数在0~1之间,区分自旋),而不是空间自然轨道(占据数在0~2之间,不区分自旋),那么除了density关键词以外还得写上pop=NOAB才行。对于G09 C.01及之后的版本就没这个问题了。
经常有人问诸如此类问题:“给Multiwfn用的波函数文件是不是必须单点任务产生的?”、“在优化后有没有必要做单点来产生?”、“能不能用优化任务直接产生的波函数文件?”。在这里明确说一下此问题。Gaussian做单点、opt、freq、NMR、polar等任务,都可以同时产生当前计算级别的波函数文件(wfn或wfx或fch)。对于opt任务,波函数文件记录的是最终结构的波函数。显然,只要你当前波函数文件里的波函数是你要想分析的,波函数文件就可以给Multiwfn用,而不用管是什么任务产生的。比如你在B3LYP/6-31G*级别下优化一个体系,并且觉得这个级别的波函数的质量对于当前分析够用了,显然就可以用这个opt任务直接得到的波函数文件做分析。如果你嫌B3LYP/6-31G*级别略low,想让波函数分析结果质量更高,那么比如可以在优化后的结构基础上再在B3LYP/def2-TZVP下算个单点任务产生波函数文件作为Multiwfn的输入文件。
对于scan、IRC任务,如果你想把每个点的wfn文件都产生出来,可以看此文《产生Gaussian的IRC和SCAN任务每个点的波函数文件的工具》(http://sobereva.com/199),由此可以做一些很有意义的分析,比如《通过键级曲线和ELF/LOL/RDG等值面动画研究化学反应过程》(http://sobereva.com/200)、《制作动画分析电子结构特征》(http://sobereva.com/86)。
(2)用GAMESS-US产生wfn文件
在$CONTRL段落中加上AIMPAC=.TRUE.关键词,然后当任务正常运行完之后,在$SCR环境变量(由rungms脚本所定义)对应的路径下的dat文件中,把----- TOP OF INPUT FILE FOR BADER'S AIMPAC PROGRAM -----"到"----- END OF INPUT FILE FOR BADER'S AIMPAC PROGRAM -----"之间的内容拷到一个文本文件里,后缀改名为wfn即可。
(3)用ORCA产生wfn文件
ORCA的输入文件里加上aim关键词,计算完成后就会在当前目录下产生与输入文件同名的wfn文件。或者在普通计算完成后,通过orca_2aim FFFFF命令把FFFFF.gbw转化为FFFFF.wfn。注意,ORCA产生wfn文件的功能可能有一定问题,文件格式不规矩,Windows版产生的和Linux版有一定差异,不同版本产生的还可能有差异。撰文时Multiwfn最新的版本可以支持ORCA 4.0的Windows和Linux版产生的wfn文件,但对于老版本产生的则可能不能正常载入,也不保证能支持未来ORCA版本产生的wfn文件。
并不是所有程序产生的wfn文件都能被Multiwfn正常载入和分析。比如有的程序产生的wfn文件格式不规矩,载入时必定导致程序崩溃。还有的程序,比如ADF,产生的wfn文件用的是Slater型函数,但Multiwfn只支持Gauss型函数,因此也没法正常分析。
3.2 wfx文件
wfx文件是对wfn文件的改进,从G09 B.01版开始引入,其介绍以及与wfn的差异在《在赝势下做波函数分析的一些说明》(http://sobereva.com/156)中有充分说明,这里不再累述。对于Multiwfn来说,用wfx文件不会比wfn文件带来什么益处。wfx比wfn最关键的改进是带有EDF字段描述被赝势代替的内核电子密度,从而对于纯粹基于电子密度的分析,赝势下的结果能和全电子基组很接近。但由于Multiwfn从3.4版开始已经自带了一套更好的EDF数据库,用其它格式波函数文件也能同样享受到EDF信息带来的好处,所以wfx文件的这个优点就不算优点了。
目前能产生wfx文件的程序还较少。在Gaussian里产生wfx文件就是把out=wfn改为out=wfx,其它完全一致。wfn和wfx文件能提供的信息对于Multiwfn来说是完全一样的。
3.3 fch文件
chk文件(checkpoint文件)是Gaussian私有的记录计算中产生的较重要信息的格式。产生chk文件很简单,Gaussian计算时在输入文件开头写上比如%chk=C:\nico\nozomi.chk,算完了之后就会产生相应的chk文件。几乎任何计算任务都会在chk里写入可以用于Multiwfn做分析的波函数信息;当然,最简单的就是单点任务了。
由于chk是二进制文件,只有Gaussian自己才能利用它,因此如果想让其它程序能够利用其中的信息,必须将它转化为文本形式的fch文件(formatted checkpoint文件)。此文件的详细介绍见前述的《高斯fch文件与wfn波函数文件的介绍及转换方法》。
fch文件、前文提到的mwfn文件和后文提到的molden、gms文件,相对于常用的wfn和wfx文件主要优势就是记录了基函数信息,从而能够利用Multiwfn的更多功能。而且它们还记录了空轨道信息,这对于涉及空轨道的分析是必须的。
对于Windows版Gaussian,把chk转化成fch文件的常规做法是启动Gaussian的图形界面,点击Utilities - FormChk,然后选择chk文件,则同名的fch文件就会产生在与chk相同的目录下。有些人心急,formchk还没转换完就把窗口关了,则得到的fch显然是不完整的,Multiwfn载入时肯定会崩溃。
对于Linux版Gaussian,应使用Gaussian目录下的formchk工具来把chk转换为fchk(fchk和fch是一码事,不同平台下默认的后缀名不同而已,Multiwfn都能识别)。比如用formchk MADLAX.chk就把此文件转化为了当前目录下的MADLAX.fchk。如果运行formchk时系统提示找不到此可执行文件,9成可能是机子里的Gaussian是菜鸟装的,步骤不合理。如果你不知道怎么搞,索性直接用绝对路径指明formchk的路径,比如/sob/g09/formchk mizuki.chk。当转换过大的chk文件的时候,可能会因为默认允许调用的内存太小导致chk转换失败,此时需要在环境变量中设置比如export GAUSS_MEMDEF=4GB,代表允许formchk调用4GB内存。
转换时应注意屏幕上的提示,如果显示了出错,则产生的fch文件载入Multiwfn时肯定会导致Multiwfn自动退出。转换时提示出错一般有三种可能
(1)chk文件莫名其妙地损坏了。把之前的chk删了,重算一遍单点再试
(2)产生chk文件的任务本身就没正常结束。应恰当修改关键词。
(3)产生chk文件用的Gaussian版本、平台和你当前用的formchk对应的Gaussian不一致。比如服务器上用Linux版Gaussian产生的chk却用Windows版Gaussian自带的formchk转换肯定会失败。最好哪里产生的chk就用哪个Gaussian自带的formchk来转换。
如果你懒得每次手动用formchk转换,那么强烈建议你把settings.ini里的formchkpath参数设为实际的Gaussian文件包中的formchk可执行文件的路径,这样使用chk作为输入文件时,Multiwfn就会自动调用formchk将之转化为fch/fchk并载入,载入成功后会自动将fch/fchk之删掉。
注意,对于后HF计算,哪怕用了density关键词,默认情况下fch文件里记录的也只是HF轨道。虽然此时也记录了后HF密度矩阵,但是Multiwfn并不会读取密度矩阵,所以此时分析的结果和HF的结果是完全一样的。类似地,做TDDFT计算时,由于默认情况fch里记录的轨道是基态DFT轨道,所以分析结果和分析基态DFT波函数也是一样的。如果想基于后HF或TDDFT自然轨道进行分析,从而得到对应后HF波函数或TDDFT激发态波函数的结果,有两种做法:
方法1:参见《在Multiwfn中基于fch产生自然轨道的方法与激发态波函数、自旋自然轨道分析实例》(http://sobereva.com/403)。此方法最推荐,很省事,而且可以生成不同类型的自然轨道。
方法2:此方法不推荐,步骤繁琐。
(1)带着density关键词做一次计算,如# MP2/cc-pVDZ density
(2)使用此关键词做一次计算# guess(save,only,naturalorbitals) chkbasis,此时%chk设的chk文件应和上一步是一致的。这一步的用处是利用chk里的密度矩阵产生自然轨道,然后转存到chk的轨道信息段落里
(3)用文本编辑器打开fch文件,把第一行里插入saveNO这个词。这样做是因为fch文件里的能量段落记录的不再是能量,而是自然轨道的占据数。当Multiwfn发现第一行有saveNO这个词,才知道要把轨道能量作为占据数来读入,否则实空间函数计算结果会错乱
然后此fch文件就可以载入Multiwfn了,如果想确认一下是否自然轨道被正确载入了,就在主功能0里把轨道信息显示出来,看到占据数确实是小数就对了。
ONIOM任务产生的fch文件给Multiwfn用于波函数分析的时候有特别需要注意的问题,见http://bbs.keinsci.com/thread-27408-1-1.html。
Q-Chem和Gaussian有一定渊源,Q-Chem也能产生fch文件,如果是>=5.0版本肯定能直接顺利载入,但如果是较老版本,需要先将settings.ini里的ifchprog设为2,否则载入的信息可能是错的。量化程序PSI4也可以产生.fchk文件,与Multiwfn完全兼容,但fchk里最高只能记录到g角动量函数(至少对于PSI4 1.2.1是如此)。
3.4 molden文件
这里说的molden文件指的是历史悠久,已经比较过时的可视化程序Molden的输入文件。虽然这个程序不好用,但molden文件倒是被很多程序所支持,成了比较通用的记录基函数信息的格式。molden能给Multiwfn提供的信息和fch一样。molden比fch有个额外好处是允许记录每个轨道的不可约表示,凡是记录了的情况,Multiwfn会读取之,在主功能0看轨道等场合会输出不可约表示。另外molden文件里明确注明了每个轨道的占据数和自旋类型,因此载入它时不需要像载入其它一些波函数文件那样需要靠程序去猜(不过Multiwfn考虑得比较周全,猜的时候也不会猜错)。
molden格式的定义也有个明显缺点,就是没有记录核电荷数的段落,而只记录了原子序数。对于全电子基组下的计算,原子序数和核电荷数是相同的。当原子用了赝势,它的实际核电荷数等于原子序数与赝势代替的内核电子数的差值。因此,如果molden文件是在赝势下计算产生的,那么Multiwfn载入后核电荷数会当成等同于原子序数,做一些涉及到核电荷的计算,比如计算分子静电势、原子电荷,结果就完全错了。为了解决这个问题,Multiwfn在读取Molden文件的时候,把[atoms]字段的第三列,也即原子序数那一类,当做实际核电荷数来读取。因此,在赝势下计算时,只要手动把原子序数那一列改成实际核电荷数,那么Multiwfn读取后就可以正常做涉及核电荷数的分析了(Multiwfn判断原子的元素是根据molden里的原子名判断的,因此原子序数改成核电荷数后,Multiwfn并不会判断错元素)。此外,ORCA程序从6.0版开始,它产生的molden文件里对用了赝势的原子在[Pseudo]字段里给出了实际核电荷数,从2024-Sep-10更新的Multiwfn开始会自动读取之,因此就不用手动做如上修改了。
molden格式还一个缺点就是支持的基函数最高角动量只到g(不过Multiwfn可以载入ORCA、Dalton产生的带h角动量的molden文件,Multiwfn也可以导出带h函数的molden文件)。
molden文件虽然有标准的格式定义,但有些地方没定义严格,再加上五花八门的量化程序往往肆意发挥,导致大多数能产生molden文件的程序产生的molden文件都是不标准的。Multiwfn虽然可以支持molden文件,但是目前版本能完美支持的只有Molpro、ORCA、MRCC、NWChem、deMon2k、Dalton、xtb、BDF产生的molden文件。对于其它情况下的molden文件,比如MOLCAS、CFOUR、PSI等程序产生的,Multiwfn载入时要么崩溃,要么载入的波函数信息有问题,通不过手册附录5说的方法的检验。对于这些Multiwfn不支持的情况,应当使用Molden2AIM(https://github.com/zorkzou/Molden2AIM)程序按照其说明设置好参数文件后,载入不规矩的molden文件,然后产生标准化后的molden文件,这样的文件就可以正常被Multiwfn所利用了。几种量化程序产生molden文件的方法如下,其它程序的产生方法见相应程序手册。
对于ORCA,任何计算都会产生gbw文件,其意义和Gaussian的chk十分类似。产生molden文件的方法是用orca_2mkl MIO -molden,就会把当前目录下的MIO.gbw转化为MIO.molden.input(是否把后缀中的多余的.input去掉无所谓),然后就可以载入Multiwfn了。orca_2mkl是ORCA程序自带的工具。如果你不想每次都手动做转换而希望Multiwfn能直接载入gbw文件,可以将settings.ini里的orca_2mklpath设为实际的orca_2mkl可执行文件路径,这样Multiwfn打开gbw文件时会自动调用orca_2mkl转换成molden文件并载入,载入完毕后会自动删除掉转换出的molden文件。
对于Molpro,把put,molden,ltwd.molden写到输入文件末尾,算完了就可以产生ltwd.molden。
对于MRCC,默认情况下任务算完了直接就会产生MOLDEN文件,将之改名,使之后缀为.molden即可被Multiwfn读取。
对于xtb,运行时加上--molden选项,任务算完了就会产生molden.input文件,可以直接被Multiwfn读取。
对于Dalton,默认情况下任务算完了之后在自动产生的.tar.gz包里有molden.inp,这就是.molden输入文件。后缀名不改也可以直接载入Multiwfn,Multiwfn发现文件名里有"molden.inp"字样就自动当作Molden输入文件读取了。
对于NWChem,在输入文件末尾加入以下内容,算完后当前目录下就会出现.molden文件
property
moldenfile
molden_norm none
end
task scf property
对于有对称性的体系,必须用"GEOMETRY noautosym"来关闭对称性。使用的基函数必须是球谐型,即使用"BASIS spherical"关键词。不满足这两点则.molden文件不能被Multiwfn兼容。
CP2K也可以产生.molden文件,如果再将晶胞平移矢量手动写入此文件,就可以通过Multiwfn做周期性体系的波函数分析,详见《详谈使用CP2K产生给Multiwfn用的molden格式的波函数文件》(http://sobereva.com/651)。
注:有些程序输出的文件格式不规矩,或者信息有误,可能在载入Multiwfn时没有崩溃、报错,但是之后分析结果错误。当你不确定输入文件里的波函数是否被正确载入了,那么建议按照手册附录5的方法来检验一下,即检查是否电子密度全空间积分值为实际电子数,以及是否每个轨道都很好满足归一化条件。如果两个测试都没问题,那么分析结果也应该没问题。
注:最常用的Gaussian程序能产生的最高级别的波函数是CCSD,如果想在更高级别波函数下分析,需要利用到PSI4和MRCC程序,详见此文《在Multiwfn中分析比CCSD更高级别波函数的方法》(http://sobereva.com/395)。
3.5 gms文件
gms文件是指的GAMESS-US或Firefly程序(原名PC-GAMESS)的输出文件,但必须手动把文件后缀名改为gms,否则Multiwfn载入时认不出这是什么文件。目前笔者只能保证Multiwfn能正确载入GAMESS-US和Firefly的HF/DFT任务在默认的NPRINT参数下的输出文件,其它乱七八糟的情况难以保证。对于几何优化,载入的是初始结构的波函数信息。gms文件能给Multiwfn提供的信息和fch、molden完全一样。
3.6 cub、grd、vti、dx文件
cub文件也叫cube文件,是Gaussian定义的一种记录格点数据的格式,非常流行,Multiwfn、VMD、ChemCraft、gview、Chem3D等诸多知名程序都支持它。Multiwfn自身也可以用主功能5计算格点数据导出成cub文件。cub文件的详细介绍见《Gaussian型cube文件简介及读、写方法和简单应用》(http://sobereva.com/125)。Multiwfn只支持三个平移矢量分别对应于X,Y,Z轴的情况,有些第一性原理程序可能会产生比如对应三斜格子的cub文件,Multiwfn虽然载入时可能不报错,但用主功能0观看等值面会乱七八糟,进行统计、处理时结果也会有问题。还有的程序产生的cub文件本来就不规矩,Multiwfn载入时会崩溃。
grd文件是DMol3程序定义的,和cub文件一样用来记录格点数据,但是grd文件一大缺点是没有记录原子坐标。所以用Multiwfn主功能0观看grd文件的等值面时会看不到实际体系的结构。产生grd文件的方法是:Material Studio里用Dmol3计算时候选上涉及格点数据的属性,比如properties里选Electron density。算完之后,在任务的目录下就有grd文件了,但在MS界面的文件列表里不显示,因为它是作为隐藏文件出现的。
vti和dx也是格点数据格式,并且不能记录原子坐标信息。vti可以由GIMIC 2.0和ParaView产生,见《考察分子磁感生电流的程序GIMIC 2.0的使用》(http://sobereva.com/491)。dx可以由比如VMD的Volmap插件(主要用于产生空间分布函数)产生。
3.7 NBO plot文件
NBO plot文件是指后缀为31~40的一系列文件,31文件记录基函数定义,32~40分别记录NBO框架中定义的各种轨道PNAO/NAO/PNHO/NHO/PNBO/NBO/PNLMO/NLMO/MO向基函数的展开系数。Multiwfn载入31后再载入32~40中的一种,就可以观看或考察相应的轨道。NBO plot文件可以用Gaussian或独立的NBO程序输出,见《使用Multiwfn绘制NBO及相关轨道》(http://sobereva.com/134)。虽然这些文件都可以提供GTF信息,但如果要考察实空间函数的话,只能用NBO(对应37)或NLMO(对应39),否则结果没意义。
3.8 pdb、xyz、mol、mol2、chg、gro、cif文件
pdb原本主要是用来记录生物大分子结构及相关信息用的格式,但目前已经成了用的最为通用的记录分子结构的格式,几乎所有分子可视化程序都支持pdb格式。pdb文件最后一列是元素名,Multiwfn载入时会优先根据元素名对原子指认元素,但有的pdb文件不规矩,没有元素名这一列,Multiwfn就只能根据pdb里的原子名来尝试判断原子的元素,但由于原子名五花八门,所以Multiwfn很容易判断错元素。所以对于没有元素名一列的pdb文件,让Multiwfn载入后一定要看一下Multiwfn在屏幕上输出的化学组成,看是否和实际一致,不一致时分析结果肯定错误。此时应自行增加元素名一列,或者把判断错元素的那些原子的原子名改为元素名,以便于Multiwfn正确判断。
xyz文件是最为简单的记录分子结构的格式,被支持得也很广泛。优点是内容非常简明,而且是自由格式,小数位数多少都随意,因此记录精度可以高于pdb。标准的xyz文件里的原子名就是元素名,但有的程序产生的xyz文件里的原子名可能并非是元素名,此时也可能造成Multiwfn读取时判断错元素。此时应该自行改成实际的元素名。
.mol和.mol2文件都是很流行的记录小分子结构的格式。此格式还明确记录了原子间链接关系(原子间形式键级)。此文件对于Multiwfn的某些依赖于原子间连接关系的计算,比如EEM电荷计算,是必须的。笔者建议用GaussView、OpenBabel等程序产生这些文件文件。
chg文件是Multiwfn自己定义的格式,用来记录原子坐标和原子电荷,每一行内容都是:[元素名] [X坐标] [Y坐标] [Z坐标] [原子电荷],比如
O 0.000000 0.000000 0.119308 -0.301956
H 0.000000 0.758953 -0.477232 0.150977
H 0.000000 -0.758953 -0.477232 0.150977
这个文件格式有特殊用处。Multiwfn的8号实空间函数是原子核电荷或原子电荷产生的静电势。因此如果载入chg文件,然后对8号实空间函数绘图,就可以考察基于原子电荷计算的静电势分布。chg文件可以自己很容易地根据已有的原子电荷手写,也可以由Multiwfn的计算原子电荷的功能(主功能7)产生。
gro文件是GROMOS格式文件,主要用于GROMACS分子动力学程序记录体系结构信息。此文件可以给Multiwfn提供坐标信息。由于此格式不记录元素名,所以元素名是靠里面的原子名去猜的,有猜错的可能。因此建议留意一下载入文件后显示的化学组成,看看有没有元素判断错的情况。
cif文件是最常用的记录晶体结构的格式。Multiwfn会恰当根据其中对称唯一原子和对称操作信息产生完整的晶胞结构,但是不支持有原子占据数不为1的情况。
Multiwfn还可以从许多文件中读取晶胞信息,从而做周期性体系的波函数分析、产生含有实际晶胞的第一性原理程序的输入文件,详见《使用Multiwfn非常便利地创建CP2K程序的输入文件》(http://sobereva.com/587)的第2节。
3.9 Gaussian和ORCA的输入、输出文件
Gaussian和ORCA的输入、输出文件都可以给Multiwfn提供坐标信息,除ORCA输出文件外还提供alpha和beta电子数信息。
Gaussian和ORCA的输入文件里的原子必须以笛卡尔坐标方式记录而不能用内坐标。
如果当前任务包含很多结构,比如是opt freq任务,则Multiwfn读取输出文件里的结构时读取的是最后一次的。对于Gaussian输出文件,可以通过settings.ini里的iloadGaugeom选择读取输入朝向的还是标准朝向下的结构。
3.10 CP2K和Quantum ESPRESSO的输入文件
Multiwfn可以直接从CP2K和Quantum ESPRESSO的输入文件里载入原子坐标信息和晶胞信息。CP2K计算产生的restart文件本身就是输入文件,因此也可以直接载入Multiwfn。
3.11 VASP的相关文件
Multiwfn支持载入VASP的POSCAR、CHGCAR/CHG、ELFCAR、LOCPOT文件。POSCAR可以给Multiwfn提供坐标、晶胞信息,CHGCAR/CHG、ELFCAR、LOCPOT还可以额外分别提供VASP计算产生的电子密度、ELF、单电子感受到的外势信息。文件名必须包含格式名,例如如果以CHGCAR格式载入,那么输入文件可以以诸如nozomi.CHGCAR、CHGCAR-maki为名。对于自旋极化计算,CHGCAR/CHG同时包含总密度和自旋密度、ELFCAR会记录对alpha和beta电子分别计算的ELF、LOCPOT会记录alpha和beta电子分别感受到的外势,Multiwfn在载入时会问你载入哪个。值得一提的是,在产生LOCPOT时,若LVHAR=.TRUE,则LOCPOT里记录的相当于静电势的负值,Multiwfn在载入时会问你是否反转符号,如果选y的话则载入的就相当于静电势;如果LVHAR=.FALSE.,则LOCPOT里记录的相当于静电势的负值+交换相关势。
4 Multiwfn的文件格式转换功能
注:如果本节介绍的Multiwfn的功能给你的研究带来了帮助,请在论文中引用Multiwfn刚启动时提示的Multiwfn程序的原文,这是对Multiwfn开发和维护最好的支持!
Multiwfn的主功能100里的子功能2可以把载入的文件转化成各种格式,目前能输出的格式包括mwfn、pdb、xyz、chg、wfn、wfx、molden、fch、wfn。还能输出47文件,这是独立的NBO程序(即GENNBO)的输入文件。还能输出带有当前原子坐标的各种主流量子化学程序(如Gaussian、GAMESS-US、Molpro、Molcas、ORCA、Dalton等等)和第一性原理程序(CP2K、Quantum ESPRESSO、VASP)的输入文件。如果Multiwfn载入的文件包含基函数信息,那么生成的GAMESS-US、Dalton和Gaussian输入文件里还可以带着初猜波函数信息。Multiwfn的产生ORCA、CP2K、PSI4输入文件的功能尤为强大和重要,见下面三篇文章的专门介绍:
《详谈Multiwfn产生ORCA量子化学程序的输入文件的功能》(http://sobereva.com/490)
《使用Multiwfn非常便利地创建CP2K程序的输入文件》(http://sobereva.com/587)
《使用PSI4做对称匹配微扰理论(SAPT)能量分解计算》(http://sobereva.com/526)
Multiwfn的这个导出文件功能使得Multiwfn可以作为格式转换器,有很多重要用处,这里举例一下,有些可能是不容易想到的:
• 把molden、gms转换成fch:这使得Gaussian以外的用户也可以使用GaussView看结构和轨道(虽然远不如用Multiwfn看轨道好)、用Gaussian自带的cubegen产生格点数据(虽然cubegen的功能只是Multiwfn很小一部分子集,而且速度、易用性远不及Multiwfn,因而几乎已没有使用价值,但cubegen唯独算均匀分布的静电势格点数据还是快于Multiwfn的)。另外,还有些程序,比如Stone的做分布多极分析的GDMA程序也必须依靠fch文件,利用Multiwfn的格式转换功能使得Molpro、ORCA、GAMESS-US等其它程序用户也能用这些程序做分析。
还值得一提的是,转化出fch后,可以用Gaussian自带的unfchk工具将之转成chk格式,这样Gaussian计算时就可以用guess=read从中读取初猜了)。
• 将Gaussian输入文件转化为其它量子化学程序的输入文件:这样Gaussian用户也可以比较容易地使用其它量子化学程序了,当然产生出的文件里的关键词都是默认的(ORCA除外),需要根据实际情况来修改,但起码比起完全从头写输入文件方便多了。对于Dalton程序,输入文件直接手写是极其困难,而借助Multiwfn就省事多了,详见《量子化学程序Dalton的编译方法和运行方式简介》(http://sobereva.com/463)。
• 把gms、fch转换成molden:这使得GAMESS-US、Gaussian用户也可以享用依赖于molden文件作为输入的程序,典型的就是那个陈旧丑陋的Molden可视化程序。
• 转化出47文件:Gaussian、ORCA等少数程序自己就可以产生NBO的输入文件47。然而很多其它程序都没这个功能,就难以做NBO分析了。然而依靠Multiwfn产生47文件的功能,GAMESS-US、MOLPRO、MOLCAS、CFOUR等其它程序的用户也能方便地做NBO分析。
• 转化成wfn/wfx文件:使得只支持wfn/wfx文件的波函数分析程序明显能支持更多的程序,毕竟很多量化程序都产生不了wfn/wfx文件。另外,对相同体系相同基组,Multiwfn载入含有基函数信息的文件格式所花的时间明显高于载入只含有GTF信息的文件,而且更占内存;对于很大体系,载入比如molden可能要花不少时间,消耗很多内存,而如果你对此体系只需要研究与实空间函数相关的问题,那么你可以把molden转化为wfx,以后再次分析这个体系时载入wfx就行了,载入耗时和消耗的内存量少得多。
• 转化成Gaussian输入文件:当手头有wfn、molden、pdb、mol等文件,你想基于其中的结构用Gaussian做计算怎么办?虽然你可以直接用文本编辑器打开文件,把里面坐标信息提出来,但显然更简单的方法是直接用Multiwfn产生Gaussian输入文件。
• 转化成pdb格式:当手头有wfn、molden、fch等文件,想用可视化程序看结构(一般可视化程序都不支持这些格式),那么你可以直接用Multiwfn转化成非常常用的pdb格式,然后再用gview、VMD、Avogadro等程序去看。
• 转化成xyz格式:Grimme的xtb、dftd3、dftd4等程序都支持xyz文件作为输入,显然只要你当前要算的体系所处的格式是Multiwfn所支持的,就可以直接用Multiwfn转成xyz然后给这些程序来算。这里笔者还提供了gjf->xyz的批量转换脚本:《一键把所有gjf文件转成xyz文件、把所有Gaussian输出文件转成gjf文件的脚本》(http://sobereva.com/530)。
• 转化成带初猜的GAMESS-US和Dalton输入文件:GAMESS-US和Dalton的SCF收敛做得远不如Gaussian等程序,有时候很简单的体系居然不收敛或者收敛到不稳定波函数上,而用Gaussian等程序去做则能得到能量更低的解,但有时候我们又非得用GAMESS-US和Dalton的一些功能,非得收敛且收敛到稳定波函数不可。此时,可以用Gaussian等程序在相同的计算级别下产生稳定的收敛的波函数,将mwfn/fch/molden文件载入到Multiwfn里,然后产生带$VEC字段的GAMESS-US输入文件,或带**MOLORB字段的Dalton输入文件,这俩字段里记录的就是其它程序已经收敛的波函数。这时用GAMESS-US或Dalton再计算立刻就能收敛稳定波函数上。而且还可以用Multiwfn产生比如UNO、定域化分子轨道等并导出上述输入文件,用于做CASSCF的初猜等目的。对于Dalton的情况我专门写了很详细的《利用Multiwfn令Dalton计算时使用其它程序产生的轨道作为初猜》(http://sobereva.com/740),给了具体例子和讲解。
Multiwfn产生的Gaussian、GAMESS-US、Dalton输入文件的计算级别是随便写的,应当根据实际情况修改。
• 转化成mkl格式:笔者专门写了篇文章说这点,见《将Gaussian等程序收敛的波函数作为ORCA的初猜波函数的方法》(http://sobereva.com/517)。
• VASP、VMD的volmap插件、Dmol3的格点数据文件转换成cub格式:VMD的volmap插件能产生的dx格式、Dmol3的grd格式、VASP的CHGCAR/CHG&ELFCAR&LOCPOT文件极少有可视化程序支持,如果你想用支持cub格式的程序去观看它们文件,那Multiwfn就派上用场了。需注意的是grd、vti文件不含原子坐标,但是cub文件又要求包含原子坐标,所以载入grd、vti后产生cub文件时Multiwfn会自动随意在cub里加入一个原子以满足cub的格式要求。
• 格点数据导出为vti格式:如《考察分子磁感生电流的程序GIMIC 2.0的使用》(http://sobereva.com/491)里所介绍的,ParaView是十分强大的可视化体数据的程序,具有许多化学领域可视化程序不具备的展现体数据的方式,如绘制流线场图等。将格点数据用Multiwfn转化成ParaView的vti格式后就可以享受ParaView的强大了。
• Gaussian输出文件转化成gjf格式:这个很有实际意义,比如几何优化任务之后要对最后一帧算单点能,就可以这样将最后一帧的结构保存成新的单点任务的gjf文件。笔者还提供了批量转化脚本,详见《一键把所有gjf文件转成xyz文件、把所有Gaussian输出文件转成gjf文件的脚本》(http://sobereva.com/530)。
还有Multiwfn用户利用Multiwfn的格式转换的功能专门写了批处理脚本,见《批量转换量子化学软件I/O文件以及提取输出信息的Shell脚本》(http://bbs.keinsci.com/thread-16231-1-1.html)。
另外值得一提的是,Multiwfn主功能6是修改波函数的功能,你可以利用其中的子功能对当前波函数尽情地根据需要进行修改(比如去掉某些原子或轨道的贡献、对波函数平移复制等),改过之后可以用Multiwfn的主功能100的子功能2来导出mwfn/molden/fch/wfn/wfx/47这些记录了波函数信息的格式。下次再做同样的分析时就可以直接载入了,而不需要再改一遍(顺带提醒一下,修改了基函数信息后,GTF信息不会直接更新。得导出比如mwfn、fch、molden文件后,再载入之,才会在文件载入的时候产生对应的GTF信息)。