使用Multiwfn考察手性体系的ECD和CPL不对称因子g以及绘制跃迁电、磁偶极矩
使用Multiwfn考察手性体系的ECD和CPL不对称因子g以及绘制跃迁电、磁偶极矩
Using Multiwfn to study ECD and CPL dissymmetry factor (g) for chiral systems and plotting transition electric and magnetic dipole moments
文/Sobereva@北京科音 2026-Jul-15
1 背景知识
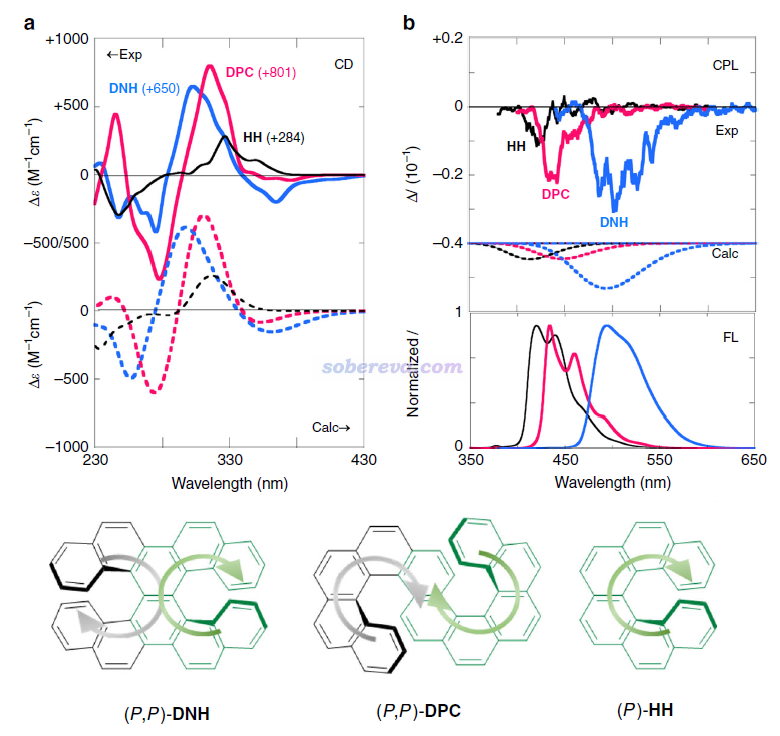
在常见的电子光谱范畴,手性体系除了像普通体系那样具有UV-Vis吸收光谱和荧光/磷光发射光谱外,还有电子圆二色谱(ECD, electronic circular dichroism)和圆偏振发光(CPL, circularly polarized luminescence)。ECD体现基态到不同激发态的电子激发对左旋和右旋圆偏振光摩尔吸收系数的差异(Δε = ε_L - ε_R),CPL体现发射态通过辐射光子退激到基态时发射的左旋和右旋圆偏振光的强度的差异(ΔI = I_L - I_R)。荧光对应的是左旋和右旋圆偏振光发射强度的总和,I = I_L + I_R。CPL和荧光发射一样通常是满足Kasha规则的,发射态是相同的。给一个典型例子,下图是Commun. Chem., 1, 38 (2018)文中给出的三种螺烯类手性分子HH、DPC、DNH的ECD谱(a图)、CPL谱(b图上侧)、荧光光谱(b图下侧),对于ECD和CPL同时给出了试验测定的(Exp)和理论计算的(Calc)谱。实验CPL光谱可以看到精细结构,是来自于振动耦合效应,而文中的理论计算没有考虑振动耦合因此没有精细结构,只是用Gaussian函数展宽成单一的峰。可以明显看出,若忽略振动耦合效应带来的精细结构的话,CPL的最大峰位置和荧光峰是完全一致的。
顺带一提,绘制ECD光谱可以参考《使用Multiwfn绘制红外、拉曼、UV-Vis、ECD、VCD和ROA光谱图》(http://sobereva.com/224)。绘制CPL光谱和获得荧光光谱一样需要先优化发射态(对普通有机分子一般是S1),然后用这个激发态的极小点结构处的它与基态间的转子强度做Gaussian函数展宽即可。
对于ECD,各个激发态都有相应的不对称因子g,一般写作g_CD或g_abs,定义如下。这是研究手性光学的文章里经常讨论的一个量,体现了特定电子激发对左旋和右旋圆偏振光吸收的选择性差异有多大。被着重报道的往往是转子强度较大,因此在ECD谱上可以观测到显著的峰的激发态的g_CD。
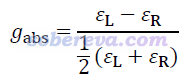
g_CD可以通过量子化学方法理论计算:g_CD = 4*R/D,其中R是转子强度、D是偶极强度。R=-|μ_tran|*|m_tran|*cosθ,其中μ_tran和m_tran分别是基态到激发态的跃迁电偶极矩和跃迁磁偶极矩矢量,θ是这两个矢量间的夹角。D=|μ_tran|^2 +|m_tran|^2。由于|m_tran|^2通常小于|μ_tran|^2几个数量级,因此也有近似关系g_CD = -4*|m_tran|/|μ_tran|*cosθ。注:很多文献里R和g_CD表达式里没有那个负号,是因为忽视了基态与激发态间的跃迁方向对跃迁磁偶极矩符号的影响。更多信息见Multiwfn手册3.21.18节。
因为ECD属于吸收光谱,因此计算g_ECD时显然要用基态的极小点结构,缺乏相关常识的话看《Gaussian中用TDDFT计算激发态和吸收、荧光、磷光光谱的方法》(http://sobereva.com/314)。
CPL的g的定义如下,一般记作g_CPL或g_lum,体现了体系发射左旋和右旋圆偏振光的倾向性程度。
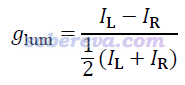
g_CPL的计算公式和g_CD完全相同,只不过由于它是发射光谱,因此要在激发态极小点结构下计算。
说一下单位的事。跃迁电偶极矩1 a.u. = 2.541746E-18 esu*cm,跃迁磁偶极矩1 a.u. = 1.85480201566E-20 erg/Gauss,必须把量子化学程序输出的以a.u.为单位的值转换成这样的单位才能再代入前述公式。相应地,算出来的R的单位是erg*esu*cm/Gauss,D的单位是esu^2*cm^2,这是cgs单位制下的具体单位。通过单位转化可以知道实际上esu*cm和erg/Gauss单位是等同的,这是为什么g=4*R/D得到的是无量纲的量。
为了便于大家计算g_CD和g_CPL,Multiwfn从2026.7.10版开始,加入了基于Gaussian和ORCA电子激发任务输出文件计算g的功能,至少兼容最常用的TDDFT的情况。Multiwfn会读取输出文件里的跃迁电、磁偶极矩并计算g,同时会把各种中间量|μ_tran|、|m_tran|、R、D、θ、cosθ全都给出,便于研究者分析影响g的各种因素。此外,这个功能还能够生成VMD的绘图脚本,使用户可以非常轻松地同时观看跃迁电、磁偶极矩的方向,很多文献里都有这种图。根据g的计算公式可知,这两种跃迁偶极矩越彼此平行,|cosθ|越接近1,g就有越大的倾向;越接近90度垂直,|cosθ|越接近0,g就倾向于越小。
Multiwfn程序和手册可以在官网http://sobereva.com/multiwfn免费下载,对Multiwfn不了解者推荐阅读《Multiwfn入门tips》(http://sobereva.com/167)和《Multiwfn FAQ》(http://sobereva.com/452)。如果在你的研究中使用了包括本文所述在内的Multiwfn的功能,请记得在文章正文里引用Multiwfn启动时提示的程序原文。
关于g_ECD、g_CD更多的计算细节请读者看Multiwfn手册3.21.18节,本文就不再展开说明了。在北京科音中级量子化学培训班(http://www.keinsci.com/KBQC)的电子激发部分有远比本文详细、深入得多的ECD和CPL的各种背景知识的讲解和详细的计算例子,推荐想学得更好更深入的人参加。下面只是给一个很简明扼要的计算例子令读者快速上手。
2 实例
P-[6]螺烯是一个非常有代表性的手性分子,也是用作本节例子的体系。所有计算,包括电子激发计算以及基态和S1态的优化,都用Gaussian 16在CAM-B3LYP/def2-SV(P)级别下完成,IEFPCM模型描述二氯甲烷溶剂环境。
2.1 ECD情况
首先计算ECD对应的g_CD和相关信息。Multiwfn文件包里的examples\excit\g_factor\目录下的TDDFT_S0geom.out是在基态极小点结构下用TDDFT算30个激发态的输出文件。启动Multiwfn,载入此文件,然后输入
18 //电子激发分析
18 //计算g因子及相关信息、可视化跃迁电/磁偶极矩
1 //这是吸收情况(ECD)
首先看到了从Gaussian输出文件中读取的跃迁电偶极矩和跃迁磁偶极矩。注意Gaussian输出文件里的跃迁磁偶极矩是标准定义的2倍,所以会被Multiwfn自动除以2
Transition electric dipole moments (a.u.):
State X Y Z norm
1 0.0201 -0.2477 0.0000 0.2485
2 0.0000 0.0000 0.1105 0.1105
3 -0.8163 2.2650 0.0000 2.4076
...略,共30个态
Transition magnetic dipole moments (a.u.):
State X Y Z norm
1 -0.1464 -0.0118 0.0000 0.1469
2 0.0000 0.0000 0.0421 0.0421
3 2.1144 -0.0396 0.0000 2.1148
...略,共30个态
接着看到以下信息,每一项的含义和单位都提示得非常清楚
Wavlen: Wavelength (nm)
|e_tran|: Magnitude of transition electric dipole moment (in 1E-20 esu*cm)
|m_tran|: Magnitude of transition magnetic dipole moment (in 1E-20 erg/Gauss)
angle: Angle between transition electric and magnetic moments (degree)
R: Rotatory strength (in 1E-40 cgs = erg*esu*cm/Gauss)
D: Dipole strength (in 1E-38 cgs = esu^2*cm^2)
g: ECD Dissymmetry factor (dimensionless)
State Wavlen |e_tran| |m_tran| angle cos(angle) R D g
1 339.5 63.17 0.272 90.01 -0.000 0.0 39.9 0.000003
2 320.6 28.09 0.078 0.00 1.000 -2.2 7.9 -0.011134
3 303.5 611.95 3.922 110.89 -0.357 856.0 3745.0 0.009143
4 288.0 106.78 0.307 0.00 1.000 -32.8 114.0 -0.011492
5 276.2 103.74 0.809 118.53 -0.478 40.1 107.6 0.014892
6 273.3 491.85 0.861 0.00 1.000 -423.6 2419.2 -0.007004
...略,共30个态
可见S1、S2的R都非常小,在ECD光谱中几乎看不到,而S3是具有较大R的最低激发态,因此是ECD谱中明显可见的峰里面波长最大的。它的D也很大,因此UV-Vis谱里面它对应的峰也非常显著。有的文章里将之算作“光学的S1”。这些光谱特征在本文第一张图里就能看到。S3的g=0.0091,和Commun. Chem., 1, 38 (2018)文中表1报道的实验值0.0092极为相符!
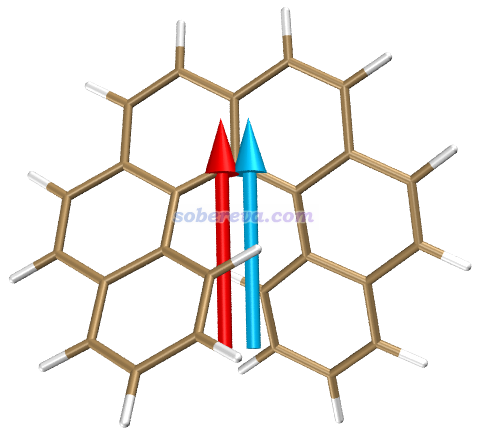
下面来观看一下S0->S3跃迁对应的跃迁电偶极矩和跃迁磁偶极矩矢量。在Multiwfn的后处理菜单选1 Generate VMD script file for visualizing transition electric/magnetic moments,然后输出产生的VMD脚本名,比如输入ECD.vmd就会在当前目录下产生ECD.vmd。然后选2 Export current structure to .pdb file,比如输入S0.pdb就会在当前目录下输出S0.pdb,里面记录了当前结构。这俩文件都已提供在了examples\excit\g_factor\目录下。
去VMD主页http://www.ks.uiuc.edu/Research/vmd/免费下载VMD 1.9.3(用其它版本后果自负)并安装。启动VMD,载入S0.pdb,并在Graphics - Representation界面里把Drawing method设成Licorice风格。把ECD.vmd拷到VMD目录下(即VMD文本窗口里运行pwd命令显示的目录),在VMD文本窗口里运行source ECD.vmd,里面定义的跃迁电/磁偶极矩和绘图命令就会被载入。之后在VMD文本窗口里运行命令emtran 3,就会看到下图,红色和青色分别是S0->S3的跃迁电偶极矩和跃迁磁偶极矩,何其方便!之前Multiwfn输出的S3的二者的夹角是110.89度,和当前箭头示意图完全对应。注:有很多文章里面的这种图把跃迁电、磁偶极矩其一或二者方向都搞反了,所以别盲目相信文献里的图!
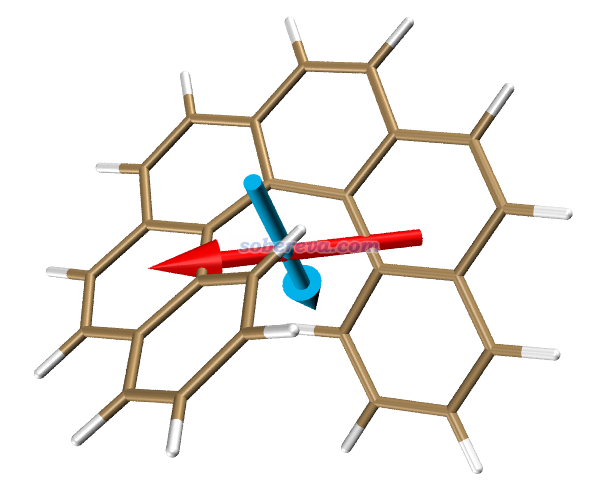
默认设置下,箭头的长度是5埃,粗细是0.15埃,箭头中心位于分子几何中心。两个箭头各自的长度、箭头的粗细、两个箭头的中心位置的平移量都可以自己用额外选项非常方便地自定义,用文本编辑器打开ECD.vmd脚本看开头部分的注释就知道用法了。
像是当前的S2中跃迁电、磁偶极矩的夹角为0度,直接画的话两个箭头会重合在一起,因此需要将二者中心稍微偏移开一些。输入emtran 2 5 5 0.15 0 0.3 0 0 -0.3 0画出来的图如下所示,跃迁电、磁偶极矩的中心分别平移了(0 0.3 0)和(0 -0.3 0)埃,因此俩箭头就错开了,看起来很清楚。
用ORCA的输出文件用于当前目的也完全可以,examples\excit\g_factor\TDDFT_S0geom_ORCA.out就是ORCA做TDDFT的输出文件例子,可以直接作为输入文件载入Multiwfn做以上分析。
2.2 CPL情况
S1态是当前体系的发射态。Multiwfn文件包的examples\excit\g_factor\目录下的TDDFT_opt_S1.out是对这个态用TDDFT做几何优化的输出文件,显然最后一次输出的跃迁电、磁偶极矩就是对应S1态极小点结构下的,由它们可以运算得到g_CPL和相关量。用这样的输出文件作为Multiwfn的输入文件,Multiwfn自动就会载入最后一次输出的跃迁电、磁偶极矩。
启动Multiwfn,载入此文件,然后输入
18 //电子激发分析
18 //计算g因子及相关信息、可视化跃迁电/磁偶极矩
2 //这是发射情况(CPL)
相同结构下,基态到激发态的跃迁(吸收过程)和激发态到基态的跃迁(发射过程),由于算符的特征原因,跃迁电偶极矩是相同的,而跃迁磁偶极矩是相反的。因此由于当前选了发射情况,Multiwfn会自动把跃迁磁偶极矩方向反转。但注意这个处理并不会影响R和g的符号(R的物理定义所致,这里不展开说)。
当前屏幕上可看到结果:
State Wavlen |e_tran| |m_tran| angle cos(angle) R D g
1 372.4 72.32 0.226 97.32 -0.127 -2.1 52.3 -0.001592
2 348.4 45.88 0.137 180.00 -1.000 -6.3 21.0 -0.011926
3 334.0 748.12 3.693 73.04 0.292 805.8 5597.0 0.005759
显然只有S1态的结果有实际意义,可见g_CPL为-0.0016。Commun. Chem., 1, 38 (2018)文中给出的实验值是-0.0009,我们的结果与之数量级相仿佛,符号也正确。Chem. Sci., 12, 5522 (2021)在与当前相同级别下计算的结果是+0.0013,大小和我们算的差不多,但符号相反,我反复研究后强烈认为是文章把符号搞反了,或者是把互为对映异构体的P型和M型螺烯的数据弄混了(这篇文章报道的实验g_CPL=0.001和Commun. Chem.那篇的-0.0009实质上相同,但符号也是相反的)。
当前g_CPL=-0.0016和S0极小点结构下S0->S1跃迁对应的g_CD=0.000003相差甚巨,符号都是相反的,这来自于S0极小点与S1极小点之间几何结构的变化,这种变化经常会导致g的符号改变。
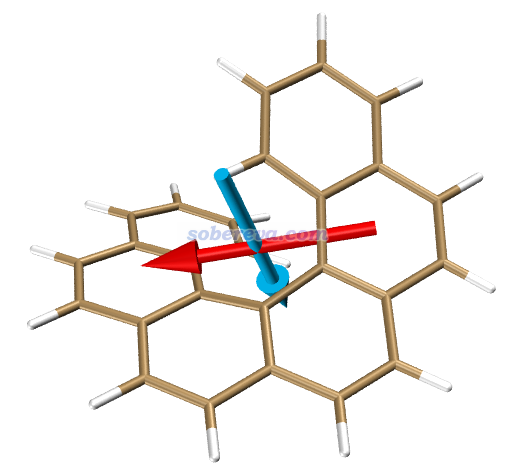
在Multiwfn后处理菜单中用选项1产生VMD作图脚本CPL.vmd,用选项2把当前结构导出成S1.pdb,然后把S1.pdb载入VMD,把CPL.vmd放到VMD目录下并用source CPL.vmd命令执行,之后输入emtran 1就可以绘制CPL过程对应的跃迁电、磁偶极矩了,依然分别对应红色箭头和青色箭头,如下所示。注:与下面的图对照可发现,Chem. Sci., 12, 5522 (2021)文中图3(e)里对应的图把跃迁电、磁偶极矩方向都弄反了。
通过本文的例子可见,Multiwfn着实令ECD和CPL的不对称g因子、相关的量的考察,以及绘制跃迁电、磁偶极矩图像的过程变得极为容易,并且不会因为研究者对很多细节理解不到位而搞错,因此十分推荐大家在对有手性的体系的电子激发研究中使用!