使用Multiwfn观看轨道概率密度
使用Multiwfn观看轨道概率密度
Using Multiwfn to visualize orbital probability density
文/Sobereva@北京科音 2024-Mar-25
0 前言
某个位置的轨道的概率密度等于轨道波函数的模的平方,对于实数型波函数来说就是轨道的平方。笔者之前有很多次被Multiwfn用户问到怎么用Multiwfn观看轨道的概率密度。传统的做法是先进入Multiwfn的主功能6的子功能26,把要考察的轨道占据数设为1,其它的都设为0,之后计算出的电子密度考察就等于那个轨道的概率密度了。考虑到这种做法步骤略麻烦,因此笔者对Multiwfn做了扩展,大大方便了考察轨道概率密度的流程,将在本文进行介绍。本文的做法适用于2024-Mar-25及以后发布的Multiwfn版本,老版本用户请去官网http://sobereva.com/multiwfn下载新版本。
如果读者不熟悉Multiwfn的话,看《Multiwfn FAQ》(http://sobereva.com/452)和《Multiwfn入门tips》(http://sobereva.com/167)。不知道波函数文件怎么产生的话,看《详谈Multiwfn支持的输入文件类型、产生方法以及相互转换》(http://sobereva.com/379)。
下面将使用苯分子作为示例,用到的波函数文件是Multiwfn程序包自带的examples目录下的benzene.fch。
1 观看轨道概率密度等的值面图
在《使用Multiwfn观看分子轨道》(http://sobereva.com/269)里详细介绍了怎么用Multiwfn非常方便快速地观看轨道波函数,没看过者务必先看一遍。观看轨道概率密度也可以在这个界面里方便地进行。启动Multiwfn,载入examples目录下的benzene.fch,然后进主功能0,在图形窗口菜单栏的Other settings里选择Choose plotting wavefunction or density,选择density然后点Return,再在窗口右下角的轨道列表里点击某轨道,或者在文本框里输入轨道序号,就看到了轨道概率密度等值面图。例如21号轨道如下
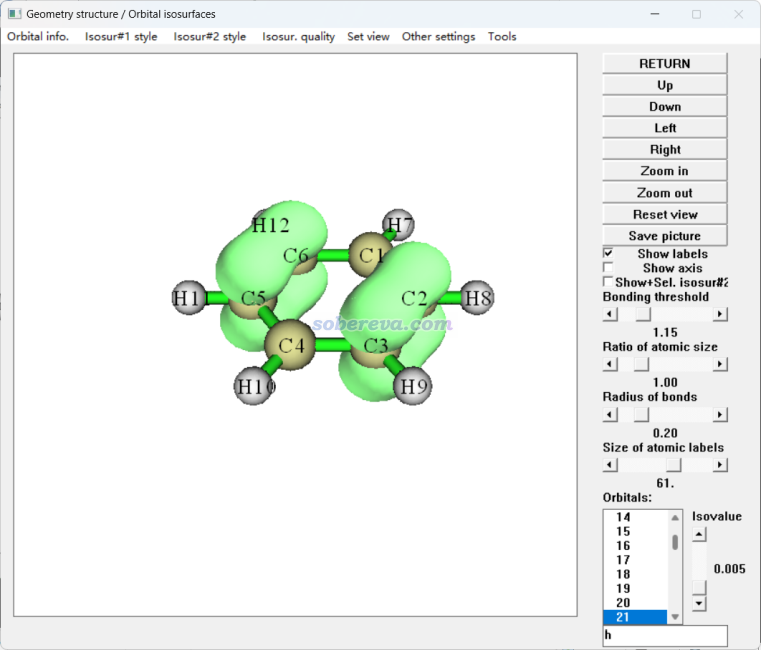
如果之后又想观看轨道波函数了,在刚才的窗口里选wavefunction即可。
2 导出轨道概率密度的cub文件
这个例子是对benzene.fch的第21号轨道计算轨道概率密度格点数据并导出为cub文件,cub文件是计算化学领域最流行的记录格点数据的格式,介绍见《Gaussian型cube文件简介及读、写方法和简单应用》(http://sobereva.com/125),导出后还可以用VMD按照《在VMD里将cube文件瞬间绘制成效果极佳的等值面图的方法》(http://sobereva.com/483)很方便地作出效果很好的图像。
启动Multiwfn,载入examples目录下的benzene.fch,然后输入
5 //计算格点数据
44 //轨道概率密度
21 //21号轨道。这个轨道是HOMO,在这里输入h也可以
2 //中等质量格点
立马就算完了。从屏幕上的提示可以看到基于均匀格点积分得到的积分值为0.999996090147538,非常接近理应的1。
现在选择2,当前目录下就出现了orbdens.cub,这就是21号轨道的概率密度的cub文件了。
3 绘制轨道概率密度平面图
benzene.fch里的分子处在Z=0的XY平面上。此例对苯分子平面上方1 Bohr的位置绘制带填色效果的等值线图。绘制平面图的更多例子和技巧看Multiwfn手册4.4节,在《量子化学波函数分析与Multiwfn程序培训班》(http://www.keinsci.com/workshop/WFN_content.html)里我还做了非常全面的讲解。
启动Multiwfn,载入examples目录下的benzene.fch,然后输入
4 //绘制平面图
44 //轨道概率密度
21 //21号轨道
2 //等值线图
[回车] //用默认的格点数
0 //设置延展距离
1.5 //1.5 Bohr
1 //XY平面
1 //Z=1 Bohr
关闭图像,然后接着输入
17 //设置显示标签的距离阈值
5 //5 Bohr
n
8 //显示化学键
14 //棕色
9 //开启等值线之间的填色效果
9 //修改填色效果
3 //设置色彩变化方式
3 //Rainbow starting from white
0 //返回
-1 //重新作图
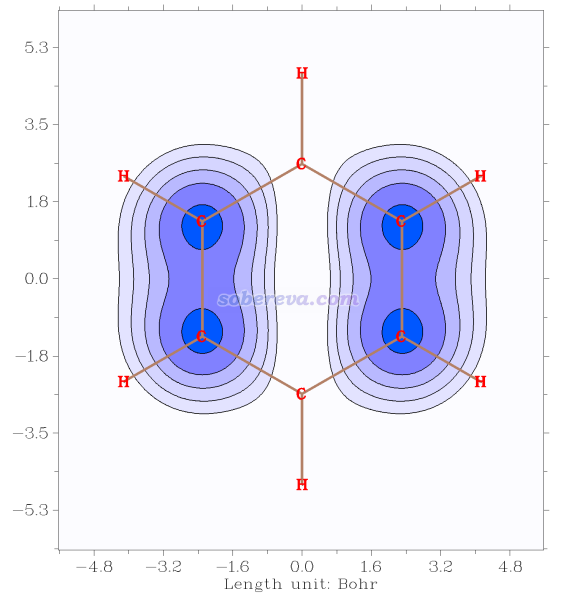
现在看到下图,效果不错。还可以用当前菜单中的选项进一步调节,如原子标签大小、坐标轴刻度等。
4 计算多个轨道概率密度的总和
基态极小点结构下的苯的HOMO是二重简并的,即20和21号轨道能量相同。这里再演示一下怎么绘制这两个轨道概率密度总和的等值面图。
启动Multiwfn,载入examples目录下的benzene.fch,然后输入
6 //修改和检查波函数
26 //修改轨道占据数
0 //选择所有轨道
0 //把所有轨道占据数设为0
20,21 //选择这两个轨道
1 //占据数设为1
q //返回
-1 //返回主菜单
5 //计算格点数据
1 //电子密度
2 //中等质量格点
-1 //观看格点数据的等值面图
在图形界面里把等值面数值设为0.005后看到下图,这便是我们想要的了。之后还可以将格点数据导出成cub文件。