CP2K做杂化泛函计算的关键要点和简单例子
CP2K做杂化泛函计算的关键要点和简单例子
Key points and simple examples of hybrid functional calculations with CP2K
文/Sobereva@北京科音
First release: 2023-Nov-7 Last update: 2024-Mar-9
0 前言
对周期性体系,杂化泛函的计算耗时众所周知远高于纯泛函。著名的第一性原理程序CP2K不光纯泛函的计算超级快,对大体系做杂化泛函计算也非常快(尽管还是远比纯泛函贵),远远快于Quantum ESPRESSO (QE)、VASP等纯粹基于平面波的程序。CP2K结合像样的基组做杂化泛函的单点计算,在一般双路服务器上算到上千原子都没问题。然而,使用CP2K做高效的杂化泛函计算有很多非常关键性的要点必须知道,远远不是像Gaussian程序那样光写个杂化泛函名字就了事的。然而,笔者经常在网上回答CP2K问题时看到很多人完全不具备这些最基本常识,胡用瞎用CP2K做杂化泛函计算,导致耗时巨高、始终出不来结果、任务崩溃、SCF完全不收敛等各种问题。因此笔者觉得很有必要写个小文将CP2K做杂化泛函计算的最关键常识和要点挨个提一下。Multiwfn程序(http://sobereva.com/multiwfn)具有极其便利的创建CP2K输入文件的功能,在本文的最后会还用Multiwfn产生C60晶体的CP2K杂化泛函计算的输入文件作为实际例子。
本文的内容至少对CP2K 2024.1,以及2023-Nov-7及以后更新的Multiwfn是适用的。
本文只是讲一些最基础的内容。我讲授的北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)的“能量的计算及相关问题”部分里讲杂化泛函的计算远远比本文具体、全面、深入得多,并列举大量经验技巧和例子,还详细介绍HSE06、CAM-B3LYP、wB97X等各种范围分离形式的杂化泛函以及revDSD-PBEP86-D3(BJ)等各种双杂化泛函的用法和要点、使用杂化泛函结合k点计算的要点等等,强烈推荐想系统、深入学习CP2K的读者参加。
1 CP2K杂化泛函计算的特性
---------
2024-Feb-18更新:以下之前写的关于k点的说法已经过时。从CP2K 2024.1版开始支持了杂化泛函计算时考虑k点,称为RI-HFXk算法,并且可以无缝结合ADMM降低耗时。此时需要以RI近似计算HF交换部分。不仅能算能量还能做优化(包括变胞)和能带计算。从第3届的“北京科音CP2K第一性原理计算培训班”开始已经加入了RI-HFXk的全面深入的介绍和具体例子,参考http://bbs.keinsci.com/thread-43683-1-1.html。
首先要知道CP2K做杂化泛函计算的一个关键不足是不支持考虑k点,因此如果被计算的是周期性体系,晶胞必须大到只考虑gamma点就足够。如果你要计算的体系的晶胞原本比较小,显然必须先扩胞到足够大才行。如果不知道扩到多大够用,严格来说需要做你感兴趣的属性的计算结果随扩胞倍数的收敛性测试。虽然CP2K能够对含有非常多原子的超胞做得动杂化泛函计算,但耗时会明显高于QE等程序对原胞在考虑k点的情况下做杂化泛函计算。
最能充分体现CP2K杂化泛函计算效率优势的情况是算一些较大分子晶体、MOF、COF等一些大晶胞体系,以及做第一性原理动力学等情况。对小晶胞体系如上所述经过扩胞后也能算,只不过不体现CP2K的长处。
CP2K显然没法用杂化泛函算能带,因为不支持k点,这种目的只能用QE等程序代替。必须用CP2K算的话,应当用CP2K支持的纯泛函里算带隙相对比较好的比如HLE17。
---------
由于杂化泛函远比普通泛函昂贵,尤其是用于几何优化、NEB、动力学等涉及到结构变化的任务。因此如果你算的问题用纯泛函就足以描述得不错、用杂化泛函不会带来明显好处的话,显然应该用纯泛函。记住,非必要甭考虑杂化泛函,纯泛函的耗时仅有杂化泛函的一个微不足道的零头。对几何优化、振动分析、NEB等高耗时任务用纯泛函,算势垒、反应能的时候用更好的杂化泛函是可以接受且常见的策略。
周期性体系的杂化泛函计算不仅耗时远高于纯泛函计算,对内存要求更是远远高于纯泛函计算。经常用CP2K做杂化泛函任务的服务器的内存容量绝对是多多益善,选择服务器的时候要注意这一点。也别天真地试图拿个人计算机将就着跑CP2K的杂化泛函计算。
CP2K做杂化泛函计算的前提是编译时必须带着libint电子积分库,否则没法算杂化泛函中HF交换项中涉及的双电子积分。CP2K的编译方法见http://sobereva.com/586。
杂化泛函用的赝势基组和赝势就用常见的给PBE等纯泛函优化的就可以。从CP2K 9.1开始,data目录下的BASIS_MOLOPT_UZH和POTENTIAL_UZH文件里还分别给出了专门给PBE0优化的赝势基组和赝势,如C的DZVP-MOLOPT-PBE0-GTH-q4和GTH-PBE0-q4,原理上来说它们用于杂化泛函计算会更理想一些(但实际未必有什么优势)。
2 库仑截断
杂化泛函计算之所以比纯泛函昂贵得多,在于计算杂化泛函的HF交换项时要计算大量的双电子积分(two-electron integral, ERI),尤其是对于周期性体系来说,不光要计算晶胞内基函数之间的ERI,还要计算中心晶胞与周围镜像晶胞基函数之间的ERI。再加上由于只能算gamma点而晶胞必然较大,原子数、基函数通常非常多,无疑要算的ERI是巨量的。
由于周期边界条件,原理上来说要计算的ERI是无穷的,显然这会导致计算无法进行。因此实际计算时必须使用库仑截断(Coulomb truncation),也就是将ERI中描述电子库仑相互作用的1/r算符截断在某个距离(截断半径),超过截断半径时1/r视为0,相应地ERI就不需要计算了,这个策略使得ERI的计算数目是有限的。如果你计算的是非周期性体系,由于本身ERI数目就是有限的,因此虽然也可以用库仑截断,但这就不是必须的了。
杂化泛函的库仑截断的半径设置显然既影响耗时也影响结果。截断半径越小,要计算的ERI越少,耗时就越低,但结果越偏离泛函原本的精确结果。显然不能为了一味地节约时间而把截断半径设得太小,否则结果会很垃圾。6埃通常是耗时和精度的较好权衡,设得明显更小会令精度有明显损失,设得更大并不会令精度有明显提升却会增加耗时不少。如果对精度要求很高也可以用8埃,设得更大就完全没必要了。
要注意像6、8埃这种程度的截断半径其实还没有大到令绝对能量充分收敛,但能量在相互求差时,由截断半径带来的系统性误差能很大程度抵消。可以认为用不同的截断半径相当于用不同的理论方法,直接对比能量的情况必须保持截断半径的设置相统一!
值得一提的是,不一定截断半径小结果就一定差,有可能截断半径造成的对ERI忽略的误差和泛函本身的误差相抵消导致恰好对某些体系某些问题的计算比用更大截断半径时结果还更好。
截断半径应当小于晶胞最短尺寸的一半以免导致得到缺乏物理意义的结果。这里所谓的最短尺寸,是(001)晶面间距、(010)晶面间距、(100)晶面间距三者中的那个最小值。也因此为了能够使用6埃截断半径,晶胞最短尺寸应当大于12埃,如果晶胞不够大则应当扩胞(即便抛开库仑截断问题不谈,单从只能计算gamma点这点来说,晶胞也不能过小)。
Multiwfn创建的杂化泛函输入文件中,若周期性设成了NONE,则不使用库仑截断,若是周期性,Multiwfn会自动加上库仑截断的设置。如果晶胞最短尺寸够大,Multiwfn会自动把截断半径设为6埃,如果不够大,则会根据当前晶胞最短尺寸自动设成能设的最大半径。
以下是库仑截断的设置方式,加入到&DFT/&XC里面。POTENTIAL_TYPE TRUNCATED代表用库仑截断的1/r算符,CUTOFF_RADIUS设置截断半径(埃)。T_C_G_DATA t_c_g.dat是默认的可以不写,这指定的是库仑截断用的gamma函数的数据文件t_c_g.dat,在CP2K的data目录下。
&HF
&INTERACTION_POTENTIAL
POTENTIAL_TYPE TRUNCATED
CUTOFF_RADIUS 6.0
T_C_G_DATA t_c_g.dat
&END INTERACTION_POTENTIAL
&END HF
3 积分屏蔽
积分屏蔽(integral screening)是指尽可能在不明显牺牲精度的前提下减少要计算的ERI的量,这对于CP2K杂化泛函计算的耗时和内存使用量有至关重要的影响!上一节的库仑截断本身就是一种积分屏蔽策略,CP2K里还有另外两种非常重要的积分屏蔽方法,也是普遍应用在Gaussian等量子化学程序中的:
(1)利用Schwarz不等式做积分屏蔽:利用Schwarz不等式关系,可以根据数目较少的二指标ERI估计出数目甚巨的三、四指标ERI中哪些数值肯定小于阈值从而可以忽略掉,这可以大幅减少ERI要算的数目(原本总共要算的ERI中占比最大的就是四个基函数序号不同的那些ERI)。阈值通过&DFT/&XC/&HF/&SCREENING中EPS_SCHWARZ设置,此值越大,被忽略的ERI数目越多、计算精度越差。默认的1E-10虽然足够保证精度,但此时太贵,没必要。通常设为1E-6就可以了,是精度和耗时的较好权衡,想要精度更好点用1E-8。
(2)结合密度矩阵做积分屏蔽:ERI对能量的贡献取决于它与特定密度矩阵元的乘积。因此哪怕ERI不是很小,但与之相乘的密度矩阵元很小,那么这样的ERI也完全不需要计算。CP2K定义了特定方法快速估计一个ERI与相应密度矩阵元乘积值的上限,如果小于上述的EPS_SCHWARZ,则此ERI就会忽略掉。这种依赖于密度矩阵的积分屏蔽默认是关闭的,如果开启则把&DFT/&XC/&HF/&SCREENING里的SCREEN_ON_INITIAL_P设为T。由于这种积分屏蔽能令耗时巨幅降低,因此我强烈建议总是开启,也因此Multiwfn产生的杂化泛函的输入文件里都自动把SCREEN_ON_INITIAL_P设成T。
基于密度矩阵做积分屏蔽有一个极其关键的要点是初猜的密度矩阵质量不能太差!然而很多人对此一无所知!默认情况下初始的密度矩阵是CP2K自动做初猜产生的,由于它和SCF收敛时的密度矩阵相差很大、质量很糙,因此此时积分屏蔽通常做得极烂,不仅可以忽略的ERI可能没忽略,还会把一些重要的ERI给忽略掉,由此可能导致KS矩阵质量很烂、SCF极难收敛。而且忽略哪些ERI是在SCF过程一开始决定好的,即便之后随着SCF迭代密度矩阵质量不断变好,也不会由此使得积分屏蔽逐步变得合理,这导致即便最后SCF收敛了,得到的能量的精度也可能很烂。因此当SCREEN_ON_INITIAL_P设T时,必须读取一个基本合理的波函数当初猜。通常是在杂化泛函计算前先用相同的基组做一个便宜的纯泛函计算,比如PBE0计算前先用PBE做一个单点计算,得到记录了PBE收敛的波函数信息的wfn文件,然后PBE0计算时用SCF_GUESS RESTART,并把WFN_RESTART_FILE_NAME指定成那个wfn文件,以读取其作为初猜。这不仅使得基于密度矩阵的积分屏蔽可以合理进行,还有一个额外的好处是由于初猜波函数质量较好(至少比默认自动产生的好得多),使得杂化泛函计算时达到SCF收敛所需的迭代次数会比较少,这进一步节约了时间(注意即便抛开ERI计算耗时不谈,杂化泛函在构造KS矩阵时花的时间也明显高于纯泛函,因为涉及到密度矩阵元与ERI的大量的乘加操作)。
一般的量子化学程序虽然也基于密度矩阵做积分屏蔽,但不是非得像CP2K这样得读取一个像样的初猜波函数,这是因为它们会根据当前最新的密度矩阵判断这一轮SCF用的ERI哪些是能被忽略的。CP2K的做法和它们不同,主要是因为CP2K倾向于让用户做incore形式的SCF而非一般量化程序一般用的direct形式的SCF,见后文。
当SCREEN_ON_INITIAL_P设T时,对于杂化泛函的几何优化、分子动力学等任务的续算,要注意光有.restart文件还不行,还必须提供上一步的.wfn文件当初猜波函数,否则也是会由于基于自动初猜的密度矩阵做积分屏蔽非常恶心而令任务无法正常跑下去。
如果你嫌麻烦而不想先做纯泛函计算产生收敛的波函数再做杂化泛函计算,那就不要写SCREEN_ON_INITIAL_P T。对于小体系、小基组特别是非周期性的计算,不基于密度矩阵做积分屏蔽时耗时也不高。而对于计算量较大的杂化泛函计算,则强烈建议不要偷这个懒而多花巨量不必要的时间。
4 ADMM
CP2K还支持其开发者独创的ADMM (Auxiliary Density Matrix Methods)方法减少要算的ERI数目从而进一步显著加快杂化泛函的计算,其原理见以下北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)的ppt。

ppt里提到的主基组就是指平时实际用的基组,诸如DZVP-MOLOPT-SR-GTH。对于不同的主基组,有一些专门构造的与之匹配的ADMM辅助基组,比如MOLOPT系列基组适合搭配的是CP2K的data目录下的BASIS_ADMM_UZH文件里的那些辅助基组,从小到大依次是admm-dz、admm-dzp、admm-tzp、admm-tz2p。admm-dzp是2-zeta结合极化函数的辅助基组,通常算是保底质量。DZVP-MOLOPT-SR-GTH主基组适合搭配admm-dzp,更好的主基组如TZVP-MOLOPT-GTH结合admm-dzp也可以接受,若结合admm-tzp精度会更好些,这都比起不用ADMM能巨幅节约计算时间和内存。辅助基组的大小与主基组越接近,ADMM给杂化泛函HF交换项带来的误差越小,但需要计算越多ERI因而越耗时。如果辅助基组和主基组设为相同的,那么ADMM不会造成任何误差,相应地也不会节约任何耗时(反倒还会增加一些耗时)。
data目录下的BASIS_ADMM文件中的FIT3、pFIT3等辅助基组都比较过时了,对元素涵盖也远不如BASIS_ADMM_UZH里的完整,因此如今不再建议使用(它们出现在不少官方例子文件和文献中,如今不要效仿)。目前版本的Multiwfn产生利用ADMM的杂化泛函的输入文件时默认设的是admm-dzp。
注意前述这些辅助基组都是对GTH赝势基组用的,千万别做全电子计算时也用它们。要想全电子计算时用ADMM,可以用(aug-)pcseg系列基组作为主基组,在《在线基组和赝势数据库一览》(http://sobereva.com/309)中介绍的BSE基组数据库中有和它匹配的(aug-)admm辅助基组,诸如pcseg-2适合搭配admm-2。pcseg是一类构造得很好的对DFT计算非常划算的基组,《谈谈量子化学中基组的选择》(http://sobereva.com/336)里简要提及了,在北京科音中级量子化学培训班(http://www.keinsci.com/workshop/KBQC_content.html)里讲基组的地方做了具体介绍。
用MOLOPT系列基组做周期性体系的杂化泛函计算几乎是一定要结合ADMM的,否则极难算得动!因为MOLOPT是完全广义收缩基组,而CP2K利用的计算源高斯函数(PGTF)之间ERI的libint库又没有专门考虑广义收缩的情况,因此造成等效的PGTF数目甚巨,ERI的数目又与PGTF数目的四次方近似成正比,数目显然多得恐怖。如果你用片段收缩基组,比如def2-SVP、6-31G**、pcseg-1、pob-DZVP-rev2之类去做杂化泛函计算,由于需要考虑的PGTF数目远比档次与之相仿佛的DZVP-MOLOPT-SR-GTH要少,因此ADMM方法不是非用不可,但不用的话耗时会显著高于DZVP-MOLOPT-SR-GTH搭配admm-dzp的情况。
不能有的原子用ADMM加速计算而有的原子不用。因此如果你用混合基组,其中有的原子用的主基组有ADMM辅助基组,有的没有,你又想用ADMM,那么没有ADMM辅助基组的那些原子的辅助基组就只能设定成其主基组。虽然这些原子没法被ADMM技术加速,但至少其它那部分原子能被加速。
前面提到的各种节约耗时的策略是彼此完全兼容的,同时利用可以最大程度节约时间。即周期性体系杂化泛函计算时一般要用库仑截断、利用Schwarz不等式做积分屏蔽、结合密度矩阵做积分屏蔽、开启ADMM。所有这些设定都会对能量产生影响,因此写文章的时候最好注明,横向对比时应统一。
启用ADMM需要在&KIND里加上BASIS_SET AUX_FIT [辅助基组名],同时增加一行BASIS_SET_FILE_NAME [辅助基组文件名]。并且在&DFT里加入以下内容
&AUXILIARY_DENSITY_MATRIX_METHOD
&END AUXILIARY_DENSITY_MATRIX_METHOD
如果用的是对角化而非OT,还需要在以上字段里加入ADMM_PURIFICATION_METHOD NONE要求不做纯化。
5 ERI的储存和内存使用量的控制
SCF有两种常见形式,incore SCF是指SCF计算一开始将所有要用到的ERI全都算出来并储存在内存里,之后SCF每一轮在构造KS矩阵时会直接从内存里读取ERI而不再重新计算。direct SCF是指SCF每一轮都重新计算要用到的ERI,这也叫on-the-fly方式计算ERI。显然,incore比direct总耗时低得多,但代价是机子的必须内存非常大,因为要存的ERI通常非常多。在CP2K的杂化泛函计算中,如果你给的内存足够存得下所有ERI,那么只有SCF第一轮的时候由于要计算所有ERI所以耗时很高,而之后SCF每一轮的耗时就很低了。如果给的内存不够储存所有ERI,那么能在内存里存多少ERI就存多少,存的那部分在之后每一轮SCF中会直接读取,存不下的ERI在每一轮SCF中会重新算,相当于介于incore和direct之间的情况。显然,做大体系杂化泛函计算时应当尽量在大内存机子上做、分配给CP2K尽可能多的内存,以让尽可能多的ERI能存到内存中、尽可能减少每轮SCF过程中要on-the-fly计算的ERI的量来节约耗时。
常有人问为什么他做杂化泛函计算时老是卡在SCF刚开始的位置不动,这是因为SCF第一轮肯定是要计算所有需要考虑的ERI,耗时显然往往很高,对大体系、大基组的情况花一、两个小时都是常事,看上去好像一直停在这里似的。如果你给的内存较少而导致大部分ERI都得on-the-fly方式去算,那么之后SCF每一轮都得花将近这么多时间,整个任务跑完可能得半天甚至一天时间。如果PRINT_LEVEL设的是MEDIUM,在SCF第一轮算完后就会输出Number of sph. ERI's calculated on the fly和Number of sph. ERI's stored in-core,前者是之后每一轮SCF都需要重新算的ERI数,后者是已存到内存中而之后不需要SCF每一轮重新算的ERI数。显然前者相对于后者越小,之后每一轮SCF比第一轮时间少得越多。
CP2K用户通常默认用popt版本,每个MPI进程在计算HF交换项时最大内存使用量由&DFT/&XC/&HF/&MEMORY里的MAX_MEMORY控制,单位为MB,扣除掉储存零碎数据的占用量外,其余都用来储存ERI。MAX_MEMORY与MPI并行进程数的乘积必须小于当前机子空余物理内存量,显然应当尽量给大以尽可能减少on-the-fly方式算的ERI量,但也不要太顶着头分配,因为进入HF交换项计算模块之前CP2K还会占一定量的内存,故要有适当余量。在SCF第一轮不断往内存里写入ERI、内存占用量不断增大的过程中,若空余内存已用完时CP2K还在继续往里存ERI,CP2K就会马上崩溃,甚至还可能导致计算机暂时失去响应。所以MAX_MEMORY应当设得恰到好处。
如果空余物理内存很有限,为了能让MAX_MEMORY能设得比较大,可以减小MPI进程数。为了不因此浪费CPU运算能力,此时可以用psmp版,此时每个MPI进程下属的OpenMP线程都可以共享这个MPI进程储存的ERI。
6 杂化泛函的定义方式
这一节简要说一下杂化泛函怎么正确在输入文件里定义。对于非周期性体系,比如用PBE0,直接在&DFT里写上以下内容即可。此时不用库仑截断而用完整的1/r算符,EPS_SCHWARZ为默认的1E-10、SCREEN_ON_INITIAL_P为默认的F。
&XC
&XC_FUNCTIONAL PBE0
&END XC_FUNCTIONAL
&END XC
显然对周期性体系绝对不能简单写成上面那样,至少也得自己去指定为使用库仑截断。结合前面的讲解,周期性体系PBE0计算的泛函定义部分一般写为下面这样。PBE0泛函由75%的PBE交换项+25%的HF交换项+100%的PBE相关项构成,所以需要恰当地定义&PBE里面的参数,并在&HF里用FRACTION指定HF成份。
&XC
&XC_FUNCTIONAL
&PBE
SCALE_X 0.75
SCALE_C 1.0
&END PBE
&END XC_FUNCTIONAL
&HF
FRACTION 0.25
&SCREENING
EPS_SCHWARZ 1E-6
SCREEN_ON_INITIAL_P T
&END SCREENING
&INTERACTION_POTENTIAL
POTENTIAL_TYPE TRUNCATED
CUTOFF_RADIUS 6.0
&END INTERACTION_POTENTIAL
&MEMORY
MAX_MEMORY 3000
&END MEMORY
&END HF
&END XC
有的泛函在&XC_FUNCTIONAL里有特定的字段,可以省得自己去定义细节参数,但HF成份依然必须自己定义,否则相当于只做了残缺的纯泛函计算,结果毫无意义。比如HF成分为10%的TPSSh泛函可以写为
&XC
&XC_FUNCTIONAL
&HYB_MGGA_XC_TPSSH
&END HYB_MGGA_XC_TPSSH
&END XC_FUNCTIONAL
&HF
FRACTION 0.1
...同上
&END HF
&END XC
更多的泛函定义方式可以直接看Multiwfn产生的相应杂化泛函的输入文件里的写法,都是严格正确的。
7 一个常见警告:The Kohn Sham matrix is not 100% occupied
做杂化泛函计算,特别是用了ADMM时,很容易在SCF开始后看到The Kohn Sham matrix is not 100% occupied警告。具体原因在https://www.cp2k.org/faq:hfx_eps_warning有介绍,这里就不多说了。对于实际计算来说,看到这个警告时,先把&QS里的EPS_PGF_ORB设为比默认明显更小的1E-12,如果计算正常,即便还有这个警告也可直接无视。EPS_PGF_ORB设得越小,越无法利用重叠矩阵和KS矩阵的稀疏性节约时间,但这对耗时的影响相对于杂化泛函计算的总耗时来说微不足道。目前版本的Multiwfn产生的杂化泛函的输入文件里自动就把EPS_PGF_ORB设成了1E-12。
对于晶胞较小的情况,本身矩阵稀疏性就差,开发者建议上来就在&QS里设MIN_PAIR_LIST_RADIUS -1从而完全不利用矩阵的稀疏性节约时间,此时绝对不会再出现那个警告,但比起用EPS_PGF_ORB 1E-12时耗时会高一些。
8 简单例子:C60分子晶体
最后,这里以C60分子晶体的单点任务作为例子演示一个标准的周期性杂化泛函的计算。必须先阅读《使用Multiwfn非常便利地创建CP2K程序的输入文件》(http://sobereva.com/587)。
此体系的cif文件和下面涉及的输入文件都可以在http://sobereva.com/attach/690/file.zip中找到。此体系结构如下所示,一共240个C原子。晶胞边长是14.07埃,对于分子晶体来说不扩胞的情况下只考虑gamma点可以接受。
启动Multiwfn,输入C60.cif的路径载入之,然后输入
cp2k
PBE.inp //要产生的PBE计算的输入文件名
0 //基于默认设置产生输入文件
现在当前目录下就有了PBE.inp,对应PBE/DZVP-MOLOPT-SR-GTH单点任务。然后接着在Multiwfn里输入
cp2k
PBE0.inp //要产生的PBE0计算的输入文件名
1 //选择理论方法
-6 //PBE0结合ADMM
0 //产生输入文件
由PBE0.inp内容可见,Multiwfn自动指定的计算设置完全合适,对应的是PBE0/DZVP-MOLOPT-SR-GTH结合admm-dzp辅助基组、6埃库仑截断、EPS_SCHWARZ 1E-6、SCREEN_ON_INITIAL_P T、开启OT。
手动把PBE0.inp中的WFN_RESTART_FILE_NAME前头的#去掉并设为PBE-RESTART.wfn,把SCF_GUESS RESTART开头的#去掉,从而使得PBE0计算时读取PBE收敛的波函数当初猜。然后根据你的机子的空余物理内存量和要用的MPI进程数设置合适的MAX_MEMORY。
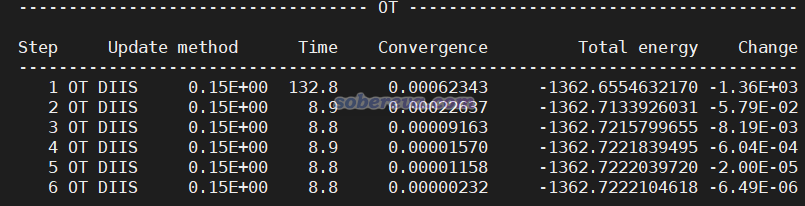
把PBE.inp和PBE0.inp放到同一个目录下,先运行PBE.inp,算完后当前目录下就出现了PBE-RESTART.wfn,然后再运行PBE0.inp。在《淘宝上购买的双路EPYC 7R32 96核服务器的使用感受和杂谈》(http://sobereva.com/653)介绍的笔者的2*7R32 512GB双路服务器上用96核并行、MAX_MEMORY设5000,用CP2K 2023.2 popt版,PBE计算花了8秒,PBE0计算花了178秒。PBE0任务的SCF迭代过程如下所示,可见第一轮耗时较高,由于当前给的内存足够储存所有ERI,因而ERI没有on-the-fly计算的,所以之后每一轮SCF的耗时远远低于第一轮。
9 总结
本文把强大的CP2K高效地做杂化泛函计算的各方面要点进行了介绍,以避免初学者对各种关键信息毫不知情而凭感觉胡用瞎用。本文的内容也充分体现出用Multiwfn产生CP2K输入文件非常容易,令研究者从复杂的输入文件编写中充分解放,但这绝不意味着CP2K的使用者就可以对关键性技术细节毫不知情,否则会各种踩坑、白浪费时间、算出无意义的结果。CP2K用户必须掌握的知识、要领远多于大部分其它计算程序。笔者开设的北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)可以使得学员完整系统透彻掌握全部这些知识。