用于非限制性开壳层波函数的双正交化方法的原理与应用
用于非限制性开壳层波函数的双正交化方法的原理与应用
Principle and application of biorthogonalization method for unrestricted open-shell wavefunctions
文/Sobereva@北京科音
First release: 2018-Nov-11 Last update: 2021-Sep-10
摘要:本文非常简要地介绍一下对非限制性开壳层波函数的alpha和beta轨道之间做双正交化的方法,并且以三重态乙醇为例介绍如何在Multiwfn中实现,使得其alpha和beta轨道最大程度匹配,从而便于讨论轨道。本文说的Multiwfn及其手册是官网上最新版本的情况。
1 相关知识
众所周知,非限制性开壳层计算(如UHF、UKS,以下简称为U)的时候alpha和beta是分别求解的,因此会产生alpha和beta这两套自旋轨道。对于自旋多重度>1的体系,以及自旋多重度为1的对称破缺态,由于自旋极化,会导致alpha和beta轨道不匹配,此时虽然alpha轨道自己是正交归一的,beta轨道自己也是正交归一的,但是alpha和beta之间不满足正交归一关系。更具体来说,第i号alpha轨道和第i号beta轨道之间重叠积分不为1,这俩轨道既可能轨道形状稍有偏差,也可能完全不同。因此,对于非限制性开壳层波函数,讨论轨道的时候必须分别去考察alpha和beta轨道,这是比较麻烦的事情。而且这种情况,体系的自旋密度是由所有占据轨道所决定的,因此没法只拿某几条轨道来讨论自旋密度。(不知道什么是自旋密度的话看《谈谈自旋密度、自旋布居以及在Multiwfn中的绘制和计算》http://sobereva.com/353)。
虽然限制性开壳层(RO)计算只会产生一套轨道,没有上述U计算的麻烦,但是相对于U,RO计算耗时高、难收敛,自旋密度、体系总能量和轨道能量也都没有U那么有意义,因此用RO回避上述问题也不是好的办法。
实际上,在U计算之后,可以做双正交化(biorthogonalization)变换来解决上述问题。双正交化的具体细节和实现在Multiwfn手册3.100.12节有详述,本文就不细说了。简单来说,双正交化是对alpha和beta轨道进行酉变换,使得alpha和beta之间完美满足或基本满足正交归一关系,同时又不破坏每种自旋轨道与其自己原有的正交归一关系。双正交化具体实现方式并不唯一,Multiwfn中的双正交化方法分成三步,经过这三步变换后,每个alpha和与之序号相同的beta轨道一般可以达到近乎完美匹配,因此之后就只需要讨论一套轨道即可(类似于RO轨道的情况)。
在双正交化过程中每种自旋的占据轨道和占据轨道会发生混合、空轨道和空轨道会发生混合(具体来说是酉变换),而不同自旋轨道间不会发生混合。这种变换不会导致体系任何可观测性质发生改变,因此如果使用双正交化后的轨道去计算体系总能量、总电子密度、自旋密度等等,结果和U计算给出的是完全相同的,在这点上类似于轨道定域化。双正交化后的轨道不再是Fock算符的本征函数,因此双正交化轨道也没有确切的轨道能量,而只能以Fock算符期望值的方式求轨道能量。
2 实例
2.1 三重态乙醇
下面我们对三重态乙醇做双正交化,来看看双正交化的实际效果和价值。首先我们先看一下UB3LYP/6-31G**下直接给出的这个体系三重态的几个分子轨道等值面图,如下所示。其中14号alpha轨道是alpha轨道中的HOMO,12号beta轨道是beta轨道中的HOMO。
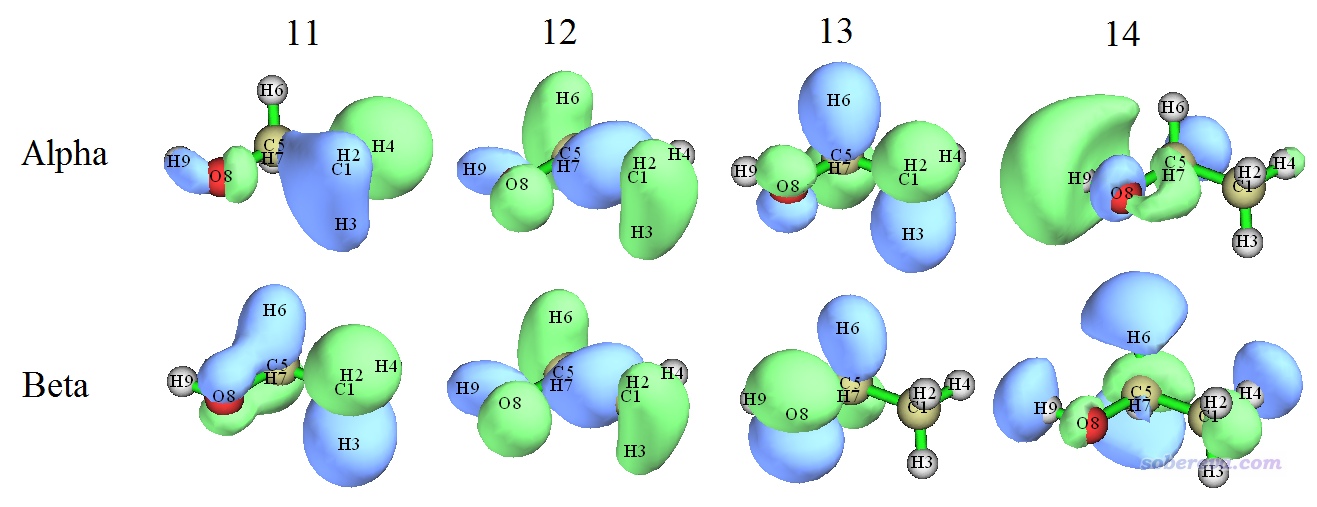
由图可见,只有12号alpha与12号beta轨道匹配很好,形状和相位都肉眼看不出显著差别,而其它alpha和beta轨道之间完全匹配不上。alpha比beta占据轨道多两个(13和14),但显然当前情况我们不可能认为体系的自旋密度等于13和14号alpha轨道对应的密度之和,因为由于其它alpha和beta占据轨道形状的不匹配,它们也对自旋密度有直接的影响。
在Multiwfn中做双正交化需要的输入文件必须包含基函数信息,具体看《详谈Multiwfn支持的输入文件类型、产生方法以及相互转换》(http://sobereva.com/379)、《Multiwfn入门tips》(http://sobereva.com/167),对于Gaussian用户就用fch文件即可。UB3LYP/6-31G**下三重态乙醇的fch文件已经在Multiwfn自带的文件包里提供了,这里我们对它做双正交化。
启动Multiwfn,然后依次输入
examples\ethanol_triplet.fch
100
12 //产生双正交化轨道
2 //双正交化过程中考虑所有轨道(对于大体系,为节约时间,可以此处选1略过空轨道之间的双正交化,此时对于此例,在最终结果中编号在14以上的alpha和beta轨道将无意义)
0 //不计算双正交化轨道能量
从屏幕上提示的信息可见双正交化分为三步进行,比如第一步输出的信息为
Doing biorthogonalization for alpha 1 to 14, Beta 1 to 12
Singular values of orbital overlap matrix:
1.0000 1.0000 1.0000 1.0000 1.0000 1.0000 1.0000 0.9999
0.9999 0.9998 0.9995 0.9992
这说明这一步是对1~14号alpha轨道和1~12号beta轨道做双正交化。下面的数值是经过双正交化后的alpha和beta轨道间的重叠积分。当前显示了12个等于或非常接近于1.0的数值,这代表这步双正交化后1~12号alpha和1~12号beta轨道已经非常完美地一一对应了。其余的轨道的双正交化会在接下来的步骤中进行。
之后Multiwfn把双正交化后的轨道波函数自动导出为当前目录下的biortho.fch文件,也把双正交化后的轨道信息导出为当前目录下的biortho.txt文件,此文件内容如下所示。
S = Singular value, E = Energy (in eV), O= Occupancy, A=Alpha, B=Beta
Orb: 1 S= 1.0000 O(A)= 1.0 O(B)= 1.0
Orb: 2 S= 1.0000 O(A)= 1.0 O(B)= 1.0
...[略]
Orb: 11 S= 0.9995 O(A)= 1.0 O(B)= 1.0
Orb: 12 S= 0.9992 O(A)= 1.0 O(B)= 1.0
-----------------------------------------------
Orb: 13 S= 1.0000 O(A)= 1.0 O(B)= 0.0
Orb: 14 S= 1.0000 O(A)= 1.0 O(B)= 0.0
-----------------------------------------------
Orb: 15 S= 1.0000 O(A)= 0.0 O(B)= 0.0
Orb: 16 S= 1.0000 O(A)= 0.0 O(B)= 0.0
Orb: 17 S= 1.0000 O(A)= 0.0 O(B)= 0.0
...[略]
通过这个文件可以非常方便地考察双正交化后的alpha轨道与序号与之相同的beta轨道的匹配程度。由于上面列出来的轨道的S(Singular value,即相当于重叠积分)都很接近1,因此这些轨道的alpha-beta匹配程度都很好,只有alpha 12和beta 12的匹配程度稍差一点,但也很接近于1.0,因此这俩轨道的图形从肉眼上应该很难分辨。上面信息里O代表Occupancy,就是占据数的意思,为了观看方便,这个文件里把O(A)=O(B)=1.0、O(A)=1.0&O(B)=0.0和O(A)=O(B)=0.0三个部分用横线做了分隔。
此时程序还问你是否现在就载入biortho.fch文件,如果选y将之载入的话,内存里的轨道就对应于双正交化后的轨道了,之后你可以进入主功能0去查看轨道图形,也可以用主功能0的图形界面左上角的Orbital info.选项去查看这些轨道的基本信息,还可以用Multiwfn中的各种功能去分析轨道特征,比如计算轨道成分、轨道动能等。由于此例没计算双正交化轨道的能量,因此在导出的biortho.fch里“轨道能量”部分记录的是上述输出的singular value值。由于当前没有双正交化轨道的能量信息,因此当前biortho.fch里的这些轨道的序号顺序并不对应于其轨道能量顺序。
这里把11~14号双正交化后的alpha和beta轨道列出:
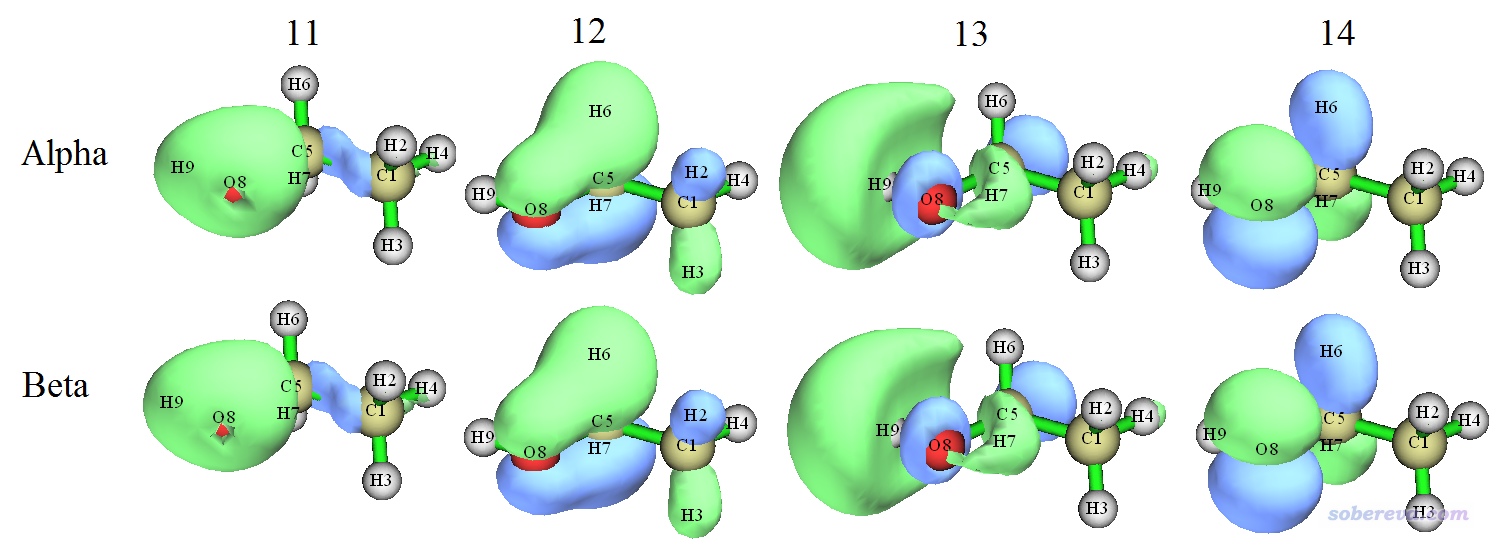
可见,双正交化后alpha和与之序号相同的beta在轨道形状上已经肉眼基本看不出任何区别了。此时,13和14号alpha轨道可以被冠以描述RO轨道时用的SOMO(单占据轨道)之名,这两个轨道对应的密度的加和正好就是当前体系UB3LYP/6-31G**级别计算的三重态的自旋密度。对比本文上面的两张图,也可以看到在双正交化前后,轨道特征往往差别不小。双正交化后的第13号alpha轨道基本正好对应原先的第14号alpha轨道,而双正交化后的第14号alpha轨道则不直接对应于之前任何一个alpha轨道,显然必定是两个或多个原先的alpha占据轨道发生了显著混合才产生出来的。
值得一提的是,乙醇的单重态基态在B3LYP/6-31G**下计算产生的HOMO和LUMO轨道图形如下
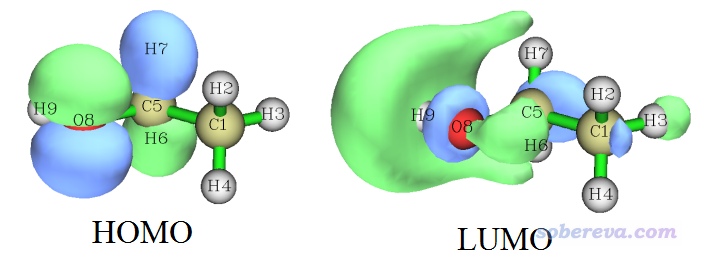
这个HOMO和LUMO分别非常像双正交化后的14号和13号alpha轨道。因此我们可以立刻明白,乙醇从单重态基态激发到最低三重态的过程,如果以轨道跃迁方式描述的话,可以近似说成是HOMO→LUMO的跃迁,因为这样跃迁后HOMO和LUMO就各有一个单电子了,这俩轨道又几乎恰好和双正交化后的两个SOMO对应。像这样重要的信息,如果我们不做双正交化的话,是没法这么容易了解到的。
2.2 三重态乙醇(计算双正交化轨道能量)
这个例子还是考察三重态乙醇,和上例不同的是这次我们也让Multiwfn产生双正交化轨道的能量,这样不仅可以讨论更多信息,而且Multiwfn会根据能量对轨道排序,讨论起来更方便。Multiwfn计算双正交化轨道的能量需要利用Fock矩阵,Multiwfn可以通过两种方式获得这个矩阵:
(1)直接基于分子轨道能量和系数矩阵反解出来Fock矩阵。通常建议用这个方式,最为省事。但是如果使用了大量弥散函数,量子化学程序可能会自动去除一些线性相关基函数,这个时候就没法用这个做法了,只能用下面的方法
(2)从特定文件里读取。可以从记录Fock矩阵的文本文件里读取,也可以从.47文件里读取(Gaussian可以直接产生),也可以从ORCA输出文件里读取,等等,详情见Multiwfn手册附录7。
启动Multiwfn,输入
examples\ethanol_triplet.fch
100
12 //产生双正交化轨道
2 //双正交化过程中考虑所有轨道
1 //计算双正交化轨道的能量,Fock矩阵通过分子轨道的能量和展开系数直接产生
y //根据能量对双正交化轨道排序
此时产生了biortho.fch和biortho.txt,后者内容如下
...[略]
Orb: 10 S= 1.0000 E(A)= -13.621 O(A)= 1.0 E(B)= -13.450 O(B)= 1.0
Orb: 11 S= 0.9992 E(A)= -13.057 O(A)= 1.0 E(B)= -11.821 O(B)= 1.0
Orb: 12 S= 1.0000 E(A)= -11.918 O(A)= 1.0 E(B)= -11.866 O(B)= 1.0
-------------------------------------------------------------------------------
Orb: 13 S= 1.0000 E(A)= -11.439 O(A)= 1.0 E(B)= -5.806 O(B)= 0.0
Orb: 14 S= 1.0000 E(A)= -0.293 O(A)= 1.0 E(B)= 2.817 O(B)= 0.0
-------------------------------------------------------------------------------
Orb: 15 S= 0.9992 E(A)= 13.876 O(A)= 0.0 E(B)= 15.431 O(B)= 0.0
Orb: 16 S= 1.0000 E(A)= 15.057 O(A)= 0.0 E(B)= 15.305 O(B)= 0.0
Orb: 17 S= 1.0000 E(A)= 18.508 O(A)= 0.0 E(B)= 19.028 O(B)= 0.0
...[略]
由于这回我们要求Multiwfn计算双正交化轨道的能量,因此这次的biortho.txt里显示了算出来的轨道能量。由于我们也要求按照能量进行排序,因此可以看到轨道序号顺序确实和轨道能量相同了。注意排序的时候是将序号相同的alpha和beta的轨道能量平均值作为依据来排序的。另外,排序是对每一个批次产生的双正交化轨道分别进行排序的(上面信息中的两条横线把所有轨道分割成了三个批次),因此比如第2批次的轨道的能量不一定全都高于第1批次的轨道。
注:常有Multiwfn用户问为什么他得到的序号相同的alpha和beta双正交化轨道的形状看起来非常相似,但能量却差了非常多。这是因为开壳层体系的alpha和beta电子的分布通常不对称,而且通常连alpha和beta电子数都往往不同,也因此alpha和beta对应的Fock算符不同。而由于轨道能量是相应自旋的Fock算符求期望得到的,因此哪怕形状精确相同、序号相同的alpha和beta双正交化轨道的能量也注定会不同。如果你就是死活非得要求相同序号的alpha和beta能量相同(就跟限制性开壳层RO计算的情况似的),那你姑且把相同序号的alpha和beta轨道能量取平均得了(虽说没有明确的物理意义),这时的轨道能量相当于轨道波函数相对于alpha和beta电子的平均的Fock算符的期望值。
当前biortho.fch里的“轨道能量”信息是双正交化轨道的真实能量了。如果我们选择y载入biortho.fch,用主功能0的Orbital info.选项查看轨道基本信息,会看到下面这些,确实轨道能量和biortho.txt里显示的相同。
1 E(au/eV): -19.26817 -524.3135 Occ: 1.000000 Typ: A
...[略]
11 E(au/eV): -0.47985 -13.0573 Occ: 1.000000 Typ: A
12 E(au/eV): -0.43799 -11.9184 Occ: 1.000000 Typ: A
13 E(au/eV): -0.42037 -11.4390 Occ: 1.000000 Typ: A
14 E(au/eV): -0.01078 -0.2933 Occ: 1.000000 Typ: A
76 ( 1) E(au/eV): -19.23884 -523.5155 Occ: 1.000000 Typ: B
...[略]
86 ( 11) E(au/eV): -0.43441 -11.8210 Occ: 1.000000 Typ: B
87 ( 12) E(au/eV): -0.43606 -11.8659 Occ: 1.000000 Typ: B
对于beta轨道,上面信息中括号里的序号是从第一条beta轨道开始计的,括号外的是从第一条alpha轨道开始计的。
看一下现在的第13号和第14号alpha轨道。如之前所述,这俩占据轨道几乎完全描述了当前体系的单电子分布,因为其它的占据的alpha轨道都有与之匹配程度极高(重叠积分接近1)的beta占据轨道故而对自旋密度贡献基本为0。这俩轨道的图形和能量如下所示
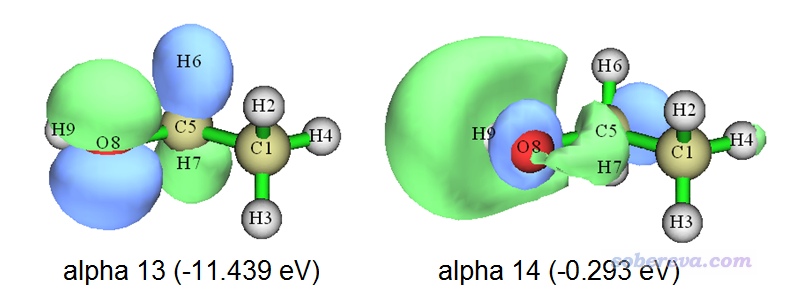
可见由于Multiwfn对轨道做了排序,当前轨道能量和轨道序号是一致的(而上例中没排序,它俩序号是反着的)。另外,如果你将这俩双正交化轨道和ethanol_triplet.fch里记录的MO13(-10.083 eV)、MO14(-0.276 eV)对比的话,会发现轨道形状相差不太大,能量也相差不算特别大,这也体现Multiwfn给出的双正交化轨道的能量是合理的。
2.3 丁烷双自由基
本节用到的fch文件可在此下载:http://sobereva.com/attach/448/C4H8-singlet.rar。如果对双自由基计算不了解,参看《CASSCF计算双自由基以及双自由基特征的计算》(http://sobereva.com/264)和《谈谈片段组合波函数与自旋极化单重态》(http://sobereva.com/82)。
通过非限制性开壳层以对称破缺方式算的自旋极化单重态体系,比如双自由基、反铁磁性耦合体系,双正交化也可以做,但是此时不可能使得每个alpha轨道都能与一个beta轨道完美相对应。比如对丁烷双自由基体系,双正交化时候的第一部分的输出是这样的
Doing biorthogonalization for alpha 1 to 16, Beta 1 to 16
Singular values of orbital overlap matrix:
1.0000 1.0000 1.0000 1.0000 1.0000 1.0000 1.0000 1.0000
0.9998 0.9998 0.9998 0.9998 0.9992 0.9990 0.9989 0.3095
可见,此时双正交化变换的效果是令alpha与beta凡是能匹配的都尽量匹配,这使得前15个轨道都有一一对应关系,重叠积分基本是1.0,但有一个alpha占据轨道就是没法与beta占据轨道相配对,重叠积分才0.3095。这样的输出信息体现在双正交化之后,我们可以就只依靠16号alpha和beta轨道来讨论体系的单电子分布以及双自由基本质特征。
顺带一提,如果想查看原本alpha和beta分子轨道之间的重叠积分,可以用Multiwfn主功能100的子功能5,具体说明详见Multiwfn手册3.100.5节。如果用这个功能里面的选项2输出对上述双自由基波函数的alpha MO和序号相同的beta MO之间的重叠积分,结果如下(以下只列出占据轨道部分)
Overlap between the 1th alpha and beta orbitals: 0.036957
Overlap between the 2th alpha and beta orbitals: -0.036727
Overlap between the 3th alpha and beta orbitals: -0.001295
Overlap between the 4th alpha and beta orbitals: 0.001066
Overlap between the 5th alpha and beta orbitals: 0.982456
Overlap between the 6th alpha and beta orbitals: -0.958735
Overlap between the 7th alpha and beta orbitals: 0.858202
Overlap between the 8th alpha and beta orbitals: -0.882210
Overlap between the 9th alpha and beta orbitals: 0.994529
Overlap between the 10th alpha and beta orbitals: -0.990360
Overlap between the 11th alpha and beta orbitals: 0.991034
Overlap between the 12th alpha and beta orbitals: -0.984323
Overlap between the 13th alpha and beta orbitals: 0.991434
Overlap between the 14th alpha and beta orbitals: -0.991413
Overlap between the 15th alpha and beta orbitals: -0.995594
Overlap between the 16th alpha and beta orbitals: 0.294899
可见,如果不做双正交化,内核轨道部分(MO 1~4)alpha-beta匹配极差,看图可知主要是因为碰巧定域在了不同原子上,这无所谓。而价层部分,7、8号轨道alpha-beta之间匹配得也不很完美,重叠积分还不到0.9,因此比如讨论自旋密度、双自由基特征时如果简单忽略它们的影响原理上并不好,而在双正交化后就仅仅需要关注第16号轨道了。请大家自行用Multiwfn基于本文提供的fch文件观看以上提及的轨道的图形。