使用Multiwfn+VMD绘制片段贡献的跃迁偶极矩矢量
使用Multiwfn+VMD绘制片段贡献的跃迁偶极矩矢量
Using Multiwfn+VMD to plot transition dipole moment vector contributed by specific fragment
1 前言
跃迁偶极矩是讨论电子激发最关键的量之一。跃迁偶极矩大,振子强度才可能大,对应的吸收/发射才可能比较强,这些基本知识在此帖都有介绍:《Gaussian中用TDDFT计算激发态和吸收、荧光、磷光光谱的方法》(http://sobereva.com/314)。量化程序给的是整个分子的跃迁偶极矩,但是我们往往想考察体系中不同部分对跃迁偶极矩的贡献,从而更好地了解跃迁的内在特征。在Multiwfn程序的主功能18里的“空穴-电子”分析模块中,可以通过Mulliken划分,把跃迁偶极矩分解为基函数的贡献,以及原子的贡献。将原子贡献加和就可以得到某个片段的贡献。如果我们把数据读入到VMD程序中,还可以在VMD中绘制箭头,直观地考察各个片段的跃迁偶极矩矢量,使各个片段对跃迁偶极矩的贡献一目了然。本文就介绍如何实现。对Multiwfn不了解者请参考《Multiwfn FAQ》(http://sobereva.com/452)、《Multiwfn入门tips》(http://sobereva.com/167)和《Multiwfn波函数分析程序的意义、功能与用途》(http://sobereva.com/184)。本文对应的是Multiwfn最新版本的情况,程序可以在主页http://sobereva.com/multiwfn免费下载。VMD使用的是1.9.3版,可以在http://www.ks.uiuc.edu/Research/vmd/免费下载。
2 原理
按照Mulliken划分,第r基函数对跃迁偶极矩的贡献写为(注意是个矢量)其中P_tran是某两个态之间的跃迁密度矩阵,后面那项是r与s基函数间的电偶极矩积分。把基函数贡献按照原子加和,就得到了原子对跃迁偶极矩的贡献。
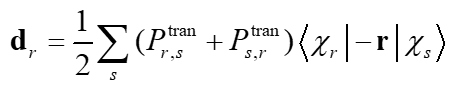
在《分子轨道成分的计算》(http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract340458.shtml)一文中,笔者就已经明确强调使用Mulliken划分时基组不能带弥散函数,否则结果缺乏物理意义。因此,按照本文方法以Mulliken划分来分解跃迁偶极矩时,做电子激发计算时候用的基组也绝对不能带弥散函数。实际上,除非里德堡激发,否则带上弥散函数对电子激发计算结果也没明显好处,基本是浪费时间,这里已经提过了:《乱谈激发态的计算方法》(http://sobereva.com/265)。
要注意,虽然体系总的跃迁偶极矩没有原点依赖性,但是片段对跃迁偶极矩的贡献往往是有原点依赖性的。换句话说,在计算时把体系进行平移,不影响总跃迁偶极矩的结果,但是可能会影响片段产生的贡献。然而原点应设在哪里,完全是任意的,因此在讨论时一定要加以注意这个问题(Gaussian计算时如果没用nosymm关键词,默认会平移体系使原点在体系原子核电荷中心位置)。之所以会有这个特征,是因为容易证明,只有最低阶的非零多极矩才没有原点依赖性,比如中性体系偶极矩没有原点依赖性,但是离子体系由于单极矩不为0,因此偶极矩有原点依赖性,因此没太大意义。类似地,对于电子跃迁问题,体系总的跃迁电荷为0,即∑[i]∑[j]P_tran(i,j)*S(i,j)=0,因此总跃迁偶极矩没有原点依赖性,而某个基函数i的跃迁电荷往往不为零,即∑[j]P_tran(i,j)*S(i,j)≠0,因此基函数、原子、分子片段对跃迁偶极矩的贡献往往是有原点依赖性的。
3 实例:偶氮苯(Azobenzene)
这里以一个简单分子偶氮苯为例,介绍一下将Multiwfn与VMD相结合,绘制各个片段跃迁偶极矩矢量的完整流程。以下是偶氮苯在PBE0/6-31G*级别做TDDFT电子激发计算的Gaussian输入文件,计算最低5个激发态。其中9/40=4必须加,否则只有较大的组态系数才会输出出来,会导致之后Multiwfn产生的跃迁偶极矩不准确(要稍微更准确的结果可以写9/40=5,不过输出文件会更大)。
%chk=C:\Azobenzene.chk
# pbe1pbe/6-31g(d) TD(nstates=5) iop(9/40=4)
pbe1pbe/6-31g(d) opted
0 1
C -0.18984100 4.51585300 0.00000000
C 1.10113800 3.99491100 0.00000000
C 1.29053000 2.61740200 0.00000000
C 0.18984100 1.75735600 0.00000000
C -1.10944900 2.28225900 0.00000000
C -1.29157500 3.65642100 0.00000000
H -0.34218800 5.59191200 0.00000000
H 1.95936300 4.66120300 0.00000000
H 2.28423900 2.17907300 0.00000000
H -1.94924600 1.59527000 0.00000000
H -2.29790100 4.06725100 0.00000000
N 0.49896400 0.37896400 0.00000000
N -0.49896400 -0.37896400 0.00000000
C -0.18984100 -1.75735600 0.00000000
C -1.29053000 -2.61740200 0.00000000
C 1.10944900 -2.28225900 0.00000000
C -1.10113800 -3.99491100 0.00000000
H -2.28423900 -2.17907300 0.00000000
C 1.29157500 -3.65642100 0.00000000
H 1.94924600 -1.59527000 0.00000000
C 0.18984100 -4.51585300 0.00000000
H -1.95936300 -4.66120300 0.00000000
H 2.29790100 -4.06725100 0.00000000
H 0.34218800 -5.59191200 0.00000000
将Azobenzene.chk用formchk转换为Azobenzene.fch。然后启动Multiwfn,载入Azobenzene.fch,之后依次输入:
18 //电子激发分析功能
11 //将跃迁偶极矩分解为基函数和原子的贡献
Azobenzene.out //Gaussian输出文件
2 //要考察的激发态。我们随便选一个,比如第2激发态
1 //分解的是跃迁电偶极矩
n //不生成AAtrdip.txt(原子-原子跃迁偶极矩矩阵)
当前目录下得到了trdipcontri.txt,其中第一部分是各个基函数对跃迁偶极矩X,Y,Z分量的贡献,第二部分是各个原子的贡献。将trdipcontri.txt拷到VMD目录下。
退到程序主菜单,然后进入主功能100的子功能2,选择导出体系的pdb文件。启动VMD,将体系的pdb文件拖入VMD main窗口载入。
将这个loadip.tcl拷到VMD目录下:loaddip.tcl。然后在VMD的文本窗口运行source loaddip.tcl命令执行之。这会将VMD目录下的trdipcontri.txt里记录的各个原子对跃迁偶极矩的贡献载入到内存(同时也输出到了文本窗口里),还定义了dip命令,用来绘制箭头表现某个片段的跃迁偶极矩,还同时定义了dipatm命令,可以一次性绘制每个原子的跃迁偶极矩箭头。
下面,我们把偶氮苯第一个苯环(原子序号1~11)、中间两个氮(原子序号12、13)和另一个苯环(原子序号14~24)分别作为三个片段来绘制它们贡献的跃迁偶极矩。用dip命令默认绘制的箭头是蓝色的,为了更显眼,先在VMD窗口里输入draw color red来让之后绘制的物体成为红色,然后在VMD文本窗口中依次输入
dip "serial 1 to 11"
dip "serial 12 13"
dip "serial 14 to 24"
这里诸如serial 1 to 11代表选择1到11号原子。dip命令绘制的箭头的圆柱部分长度对应片段贡献的跃迁偶极矩大小,箭头方向表示跃迁偶极矩矢量方向,箭头的中央位置对应选中的片段的几何中心。每次用dip命令后,在VMD的文本窗口中还会把这个片段的几何中心以及对跃迁偶极矩贡献的X,Y,Z分量直接输出出来。
注:被绘制的片段里的原子序号可以不连续。例如运行dip "serial 1 5 to 8 11 to 14 18",会对由1、5、6、7、8、11、12、13、14、18号原子构成的片段进行绘制。
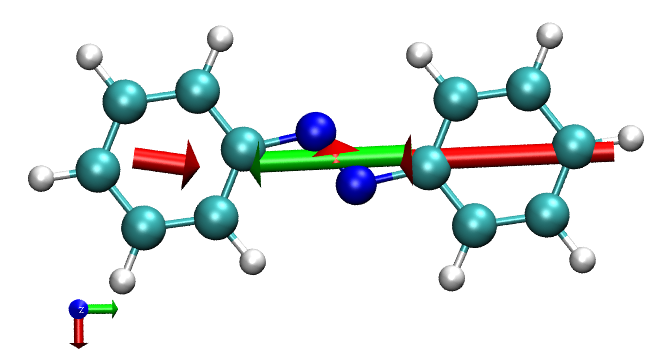
在文本窗口输入color Display Background white把背景设成白色,然后进graphics-representation,把Drawing method设为CPK。笔者建议用正交视角观看当前图像,以免因近大远小造成视觉误差,故选Display-Orthographic。最终看到的效果如下所示(左下角的坐标轴的红、绿、蓝分别对应X,Y,Z正方向)
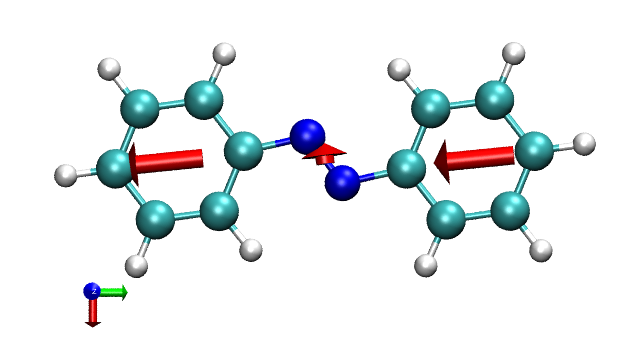
Gaussian输出的此体系S0->S2的总跃迁偶极矩,以及VMD文本窗口中看到的苯环1、N2、苯环2的贡献分别为(a.u.)
Total:0.1155 -2.8868 0.0
苯环1:0.14262 -1.43288 0.0
N2: -0.16948 -0.02138 0.0
苯环2:0.14262 -1.43288 0.0
可见跃迁偶极矩主要是Y轴负方向的。将定量数据和图像结合观察,可见两个苯环起到了最主要的贡献。而N2对Y方向贡献甚微,在X方向有一点微小贡献。
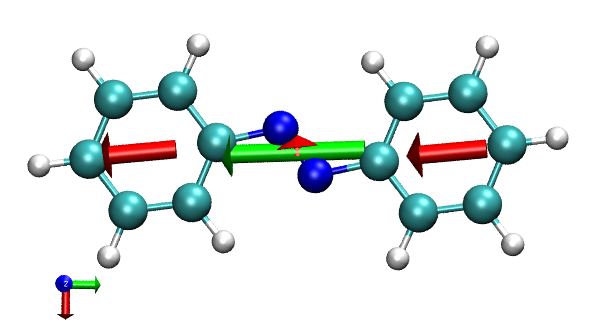
我们也可以同时把体系总的跃迁偶极矩箭头绘制出来。为了令其颜色与片段的区分,我们先输入draw color green,然后再输入dip all,就得到了下图,绿色箭头描述体系总跃迁偶极矩,其具体数值也显示在了文本窗口中。
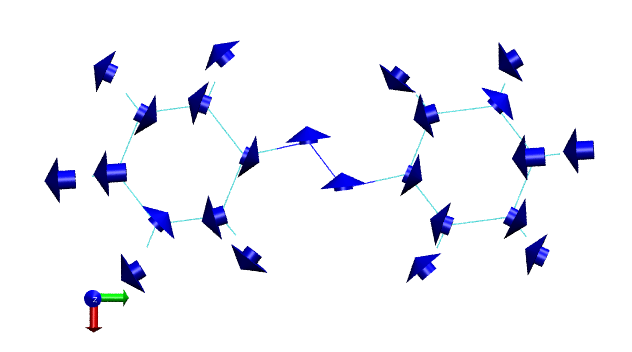
要想删除所有已绘制的箭头,输入draw delete all。下面我们输入dipatm,看看各个原子贡献的跃迁偶极矩的箭头。由于原子贡献的往往比较小,为了避免箭头被原子球遮挡,drawing method我们改成line。
前面提到,片段对跃迁偶极矩的贡献一般是有原点依赖性的。为了展示这一点,对前面的.gjf文件里每个原子Y坐标都加上6埃,并且用了nosymm关键词避免自动被Gaussian平移。重复之前的作图步骤,这次得到的图像如下
和之前的图对比,我们看到绿色箭头,即总跃迁偶极矩并没有因为体系的平移而发生改变,但是两个苯环产生的贡献却与之前截然不同,这是因为这两个苯环的跃迁电荷都不为0(用Multiwfn的空穴-电子分析界面的子功能6可以输出每个原子的跃迁电荷,按照片段进行加和,得到的两个苯环的数值分别为0.2116和-0.2116)。而N2部分跃迁电荷恰为0,因此它贡献的跃迁偶极矩和平移前一样,依然是-0.16948 -0.02138 0.0。
4 其它
上面的例子是将基态与激发态之间的跃迁电偶极矩分解成原子/片段的贡献,也可以将跃迁磁偶极矩以相同的方法进行分解并绘制,只要在Multiwfn问你分解哪种跃迁偶极矩的时候选Magnetic即可。另外,2020-Dec-25及之后更新的Multiwfn也支持对两个激发态之间分解它们的跃迁电偶极矩,让你输入激发态序号的那一步输入两个相应的激发态序号即可。
顺带一提,如果想令VMD中绘制的箭头长一点或者短一点,可以修改loaddip.tcl当中的这一部分:
set begx [expr $cenx-$fragdx/2]
set begy [expr $ceny-$fragdy/2]
set begz [expr $cenz-$fragdz/2]
set endx [expr $cenx+$fragdx/2]
set endy [expr $ceny+$fragdy/2]
set endz [expr $cenz+$fragdz/2]
将$fragdz改为比如2*$fragdz,就可以让箭头长度变为之前两倍。箭头的粗细可以通过修改draw cylinder这一句的radius后面的值来调整。