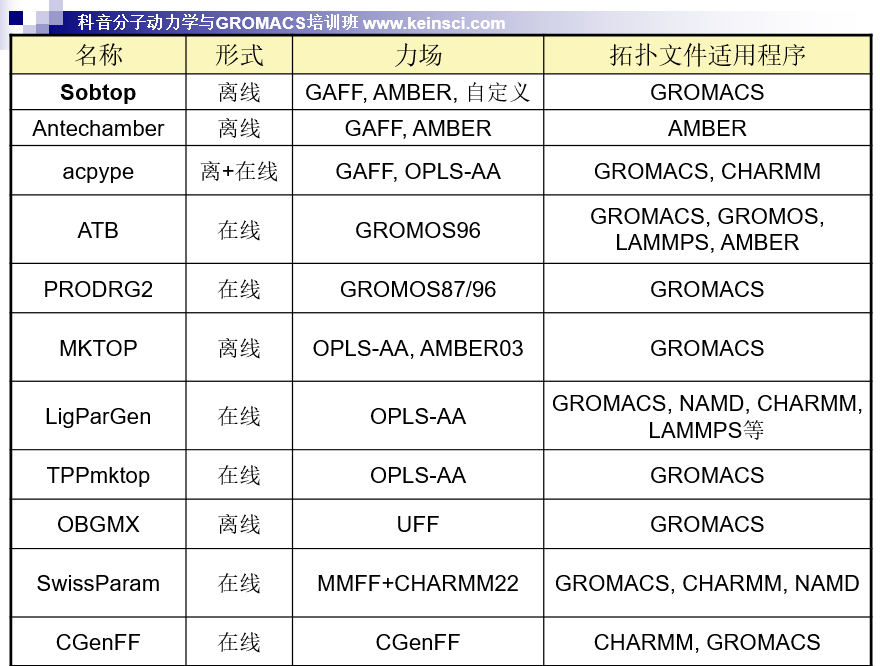
几种生成有机分子GROMACS拓扑文件的工具
几种生成有机分子GROMACS拓扑文件的工具
Several tools for generating GROMACS topology files of organic molecules
文/Sobereva@北京科音
First release:2014-Dec-9 Last update: 2024-Nov-5
本文把各种可以产生GROMACS拓扑文件的工具进行汇总。具体细节和实际使用例子笔者在北京科音分子动力学与GROMACS培训班里会详细讲(http://www.keinsci.com/workshop/KGMX_content.html)。
值得一提的是,这些程序有很多不能产生原子电荷,或者产生的原子电荷质量较差。RESP电荷是最适合分子动力学模拟使用的电荷之一,AMBER、GAFF、GLYCAM等力场将之作为御用的原子电荷,将RESP电荷与UFF力场结合使用也是得当的。Multiwfn是计算RESP电荷最简单、最好、最灵活而且完全免费的工具,原理、用法和实例见《RESP拟合静电势电荷的原理以及在Multiwfn中的计算》(http://sobereva.com/441)、《计算RESP原子电荷的超级懒人脚本(一行命令就算出结果)》(http://sobereva.com/476)、《RESP2原子电荷的思想以及在Multiwfn中的计算》(http://sobereva.com/531)。另外,OPLS-AA力场很适合结合1.2*CM5电荷使用,用Multiwfn的主功能7的子功能16直接就能计算出CM5电荷,将之手动乘以1.2即是1.2*CM5电荷,可以用Gaussian、ORCA等程序产生的fch/wfn/wfx/molden等格式作为Multiwfn的输入文件,详见《详谈Multiwfn支持的输入文件类型、产生方法以及相互转换》(http://sobereva.com/379)。如果你完全不会用Gaussian的话,可以直接用这个傻瓜式脚本,一行命令就能算出来:《计算适用于OPLS-AA力场做模拟的1.2*CM5原子电荷的懒人脚本》(http://sobereva.com/585)。另有专门结合ORCA用的傻瓜式脚本,见《ORCA结合Multiwfn计算RESP、RESP2和1.2*CM5原子电荷的懒人脚本》(http://sobereva.com/637)。
• Sobtop(http://sobereva.com/soft/Sobtop)
这是我开发的GROMACS拓扑文件产生工具,主要产生GAFF/GAFF2、AMBER力场的拓扑文件,但由于其力场库可以自行非常方便地修改和扩充,因此Sobtop本质上是完全普适、通用的。Sobtop可谓是最理想、最灵活、最易用的产生GROMACS拓扑文件的工具。此程序用起来超级简单,什么额外的程序以及特殊的运行环境都不需要装,解压即用。Sobtop使用极其方便,照着屏幕上的提示敲几下键盘,itp、top和gro文件就产生了,另外也可以要求产生rtp文件。Sobtop的主页有非常详细的产生各类体系拓扑文件的例子,并给出了详细的相关要点的说明。从例子中你会体会到Sobtop的设计特别注重兼顾便利和灵活,初级用户会体会到它极其便利,而高级用户则会体会到通过Sobtop构建复杂体系拓扑文件特别灵活好用。
Sobtop可以产生任意体系的拓扑文件,有机和无机小分子、过渡金属配合物、聚合物、共价团簇、晶体、二维材料等等全都可以产生,孤立体系和周期性体系都支持。Sobtop可以用于包含任意元素的体系,那些GAFF/GAFF2/AMBER不支持的元素可以自动让Sobtop用UFF的,或者自己定义额外原子类型。对于GAFF/GAFF2/AMBER力场里缺失的成键相关参数,可以直接让Sobtop基于量子化学程序产生的Hessian矩阵通过不同方法自动算出来,也可以自己把其它方式得到的(如自己单独拟合、文献中找的)添到力场库文件里让Sobtop直接用。Sobtop运行效率特别高,甚至对于几千原子体系的拓扑文件都可以搞。
Sobtop极度灵活,提供许多不同的指认原子类型的方式,可以自动指认GAFF/GAFF2/AMBER原子类型,也可以手动指认,也可以自定义判断规则让Sobtop结合实际化学环境和成键关系判断,等等。Sobtop还提供了丰富的指认力场成键相关参数的方式,比如可以都用力场库文件里的,都用直接基于Hessian算出来的参数,或者一部分作为刚性(里面的参数都基于Hessian算出来的)而其余部分作为柔性(参数用力场库里的,可以考虑二面角的可旋转性),等等。Sobtop还可以调用Multiwfn或OpenBabel自动指认EEM、Gasteiger、MMFF94原子电荷,还可以载入Multiwfn计算RESP/RESP2等电荷产生的chg文件来获得原子电荷。Sobtop的设计在各方面都特别贴心,比如在产生的拓扑文件里非常详细地注释各个力场项的参数来源、是否是缺失的,在自己手动改和补参数时很方便。
有了Sobtop,下面介绍的acpype就没有任何使用价值了。acpype只适合产生GAFF能描述的那些有机和极少数无机体系,有特殊成键方式、有GAFF不支持的元素、周期性体系都没法搞;对很大体系耗时非常高;容易出现莫名其妙又不好解决的报错;还得有Python运行环境、安装臃肿的AmberTools...
• acpype
这是一个Python脚本,可以在https://github.com/alanwilter/acpype下载。使用前必须在机子里安装AmberTools(免费),acpype会调用其中的Antechamber先产生Amber格式的拓扑文件然后再转成GROMACS的。acpype用法很简单,要处理xxx.mol2就执行./acpype.py -i xxx.mol2,算完后会新产生一个xxx目录,里头有_GMX后缀的.gro、.itp、.top,直接在GROMACS里用即可。默认情况下,产生的拓扑文件是基于GAFF力场的,另外也会输出_OPLS后缀的基于OPLS力场的文件,但属于实验性质不建议用。.mol2文件用常用的GaussView就可以产生(但必须确保在gview里看到的分子结构中没有诡异的成键方式),也可以通过OpenBabel或Antechamber将其它格式转成.mol2。默认情况下acpype分配的原子电荷是Antechamber产生的AM1-BCC,虽然能用,但明显不如RESP/RESP2电荷理想。建议大家按前述做法用Multiwfn计算出RESP或RESP2电荷,自行写入分子拓扑信息的[atoms]的原子电荷那一列。
如果你懒得为了用acpype而装臃肿庞大的AmberTools,可以用在线版http://bio2byte.be/acpype/,不过可能排队要排很久。所以如果你要快速处理较多小分子,还是建议用离线版acpype。
对于稍大的体系,用独立的acpype时强烈建议加上-c user选项以避免acpype自动算AM1-BCC原子电荷,否则在处理期间acpype所调用的Antechamber会先调用SQM程序用半经验方法进行优化然后再算这个电荷。然而SQM不仅慢,做优化还容易不收敛,导致半天也无法成功产生拓扑文件。更何况最后也是要替换为Multiwfn算的RESP/RESP2电荷,故计算AM1-BCC电荷没实际意义。同理,用在线版acpype也是建议把charge method设为user。
• Automated Topology Builder(ATB,http://atb.uq.edu.au):生成GROMACS拓扑文件的在线工具,可以生成基于ATB开发者自行修改的GROMOS96 G54A7力场(扩充了原子类型)的GROMACS或GROMOS程序的拓扑文件。比PRODRG2更先进更可靠,解决了PRODRG2没法自动确定质子化态和charge group指认不准的问题以保证原子电荷可靠,并且能利用对称性保证电荷等价,另外没有PRODRG2那样对于每日提交的数目有限制。ATB虽然设计得不错,可以作为产生小分子GROMOS力场的GROMACS拓扑文件的首选,但它生成的原子电荷、确定的参数的可靠性也只是一般,不能保证总是很理想。如果要做很严谨的模拟研究,还是建议自行计算RESP电荷并且手动检查ATB给出的拓扑文件的合理性,有必要时适当调节。ATB网站上也有个分子库,包含巨量事先搞好的小分子的参数和拓扑文件,其中有的是别人之前提交过的分子,有的是经过专人手工处理过的分子,显然后者可靠程度更高。对于比较常见的分子,提交到ATB之前建议先搜索一下分子库,如果已经有的话就直接用。
• LigParGen(http://zarbi.chem.yale.edu/ligpargen/):生成GROMACS, NAMD, CHARMM, LAMMPS等程序OPLS-AA力场的拓扑文件的工具。毕竟这是OPLS力场开发者自己搞的,因此原则上比MKTOP、TPPmktop更靠谱。可以通过SMILES字符串、.mol、.pdb文件进行输入(用pdb格式时老是报错,我建议用.mol),原子上限200个,可以顺带着让服务器对分子在OPLS-AA力场下做几何优化。分配的原子电荷是1.14*CM1A或1.14*CM1A-LBCC,前者是把CM1A电荷数值乘上1.14得到的,后者是在1.14*CM1A基础上再引入LBCC校正得到的,在J. Phys. Chem. B, 121, 3864 (2017)中已证明这两种原子电荷结合OPLS-AA模拟有机体系凝聚相可以得到不错结果。此服务器还可以输出PQR文件。
本文开头所述的Multiwfn可以计算的1.2*CM5电荷比LigParGen直接给出的电荷在多数情况下更适合做动力学模拟,特别是对于密度、蒸发焓的精度方面而言,这点在J. Phys. Chem. B, 121, 3864 (2017)的测试中充分体现了。而且对于一些分子,LigParGen给出的原子电荷加和不精确为0,而是比如0.0001,这导致此类分子数目较多时体系总电荷对整数有不可忽视的偏离,对模拟造成不良影响。因此建议大家将LigParGen给出的itp文件里的原子电荷部分替换为Multiwfn得到的1.2*CM5电荷。
• MKTOP:是个Perl脚本,可以在https://github.com/aar2163/MKTOP/blob/master/mktop.pl下载。支持OPLS-AA和AMBER03力场,电荷没法自动生成而必须自己提供。使用之前需要确保已经安装了Perl运行环境,并且要把mktop.pl脚本开头的GROMACS拓扑文件目录改成当前机子里实际的,比如$gromacs_dir="/sob/gmx2018.8/share/gromacs/top";。之后可以比如这样运行:./mktop.pl -i new.pdb -o new.top -ff opls -conect no。这代表处理当前目录下的new.pdb,产生名为new.top的拓扑文件,使用OPLS-AA力场,并且不使用.pdb里的CONECT字段记录的连接关系而是根据原子之间距离猜测连接关系。之后需要自行把原子电荷填到new.top的[atoms]字段里。我发现有时候此脚本指认的OPLS-AA原子类型是错误的,所以建议按照力场目录下的atomtypes.atp仔细看看指认的原子类型到底是否合理。
注:以前MKTOP是有在线服务器的(http://www.aribeiro.net.br/mktop/),但是如今已经失效。
• OBGMX(http://software-lisc.fbk.eu/obgmx/):生成UFF力场的gromacs拓扑文件的在线工具。由于UFF几乎涵盖整个周期表,因此不光有机分子,也可以使得Gromacs能够处理无机物、周期性体系,比如MOF。由于UFF的力场形式和Gromacs所支持的不完全兼容,此程序给出的参数实际上是对原UFF力场的近似。个别原子类型识别可能有误。电荷必须自己提供。
2020-Dec-26注:此在线服务器目前已无法使用,但可以下载离线程序使用,见http://bbs.keinsci.com/thread-21015-1-1.html。
• SwissParam(http://swissparam.ch):输入有机小分子mol2文件,生成用于CHARMM/NAMD和GROMACS模拟的拓扑、参数文件。力场参数基于MMFF,但只保留谐振项部分,因此只是MMFF的近似。原子电荷通过MMFF方法获得。范德华参数采用CHARMM22中最接近的原子类型。这样的参数比较粗糙,有优化的余地。
• ztop:计算化学公社论坛(http://bbs.keinsci.com)上钟成开发的拓扑文件产生工具,请在论坛首页搜索框里搜索ztop看他发的相关帖子了解此程序的特点和使用。
• CGenFF(https://cgenff.com):能够生成CGenFF力场的GROMACS拓扑文件的在线工具,学术用户必须用edu邮箱才能注册。
以下在线程序已失效:
• PRODRG2(http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg):历史非常悠久很有名的生成Gromacs拓扑文件的在线工具。只支持GROMOS87/96力场,生成gromacs和其它一些程序的拓扑文件。原子电荷是根据基团指认的,如果基团识别不对那么电荷也不可靠。有在线版和离线版。可以选择自动优化分子结构。此工具在J. Chem. Inf. Model., 50, 2221 (2010)被曝光往往不能正确指认charge group,导致原子电荷分配也很不合理。而且又由于有了更好的ATB,因此如今强烈不推荐使用
• CGenFF(https://cgenff.paramchem.org):先注册,上传有机小分子的mol2文件,即可生成基于CGenFF力场的CHARMM的拓扑文件。如果mol2是gview建的,一定要事先把里面的Ar替换成ar。上传文件的时候不要选Guess bond orders from connectivity和Include parameters that are already in CGenFF。如果文件处理正常,会立刻产生str文件,点击其链接之后把里面内容拷到比如Actos.str里面。进入网页里的More Info & Tools - Utilities,点击GROMACS conversion program,可下载把str文件转换为GROMACS格式的Python脚本cgenff_charmm2gmx.py。对于CentOS 7.2,应运行yum install numpy和yum install python-networkx把这个脚本所需的包装上。去http://mackerell.umaryland.edu/charmm_ff.shtml#gromacs里面下载用于GROMACS的CHARMM36力场文件,解压得到比如charmm36-nov2016.ff文件夹,将它和Actos.str、cgenff_charmm2gmx.py都放到当前目录,另外也把此文件夹拷到gromacs的top目录下。假设Actos.str里RESI后面的词是Molecu,最初上传的是Actos.mol2,则运行./cgenff_charmm2gmx.py Molecu Actos.mol2 Actos.str charmm36-nov2016.ff。在当前目录会产生molecu.top、molecu.prm、molecu.itp、molecu.pdb(从mol2转换过来的),适当调整itp和top里的分子名,调整itp里的残基名和结构文件相对应后,即可用于模拟。str文件里的力场参数有penalty指标,数值越大说明此参数可靠性越低,详见网页里的说明。
• TPPmktop(http://erg.biophys.msu.ru/tpp/):在线工具,提供pdb文件,能产生OPLS-AA力场参数的GROMACS拓扑文件。速度比较快,但得到的拓扑文件里有时候会缺参数,或者出现额外的原子类型,需要再手工处理(需要引入额外的力场文件,但是笔者发现在网上下载不到)。此服务器有时候有其它任务在跑,此时无法提交任务,只能等过一阵服务器没任务时再提交。
另外说两个有关的程序
• YASARA AutoSMILES Server(http://www.yasara.org/autosmilesserver.htm)是在线版程序,离线的话得买YASARA(建模+可视化+动力学模拟工具)。可以计算AM1-BCC电荷、指认amber94/96/99力场参数。但是产生的文件只能用YASARA View打开,也就是说,拓扑文件相当于YASARA专用的。
• RED(RESP ESP charge Derive,http://q4md-forcefieldtools.org/REDServer-Development/):专门生成RESP电荷的,也能搞力场参数。页面做得不是一般的糟糕,结构十分混乱,令我摸不到头绪,因此笔者也没怎么仔细研究。由于Multiwfn已经完美支持RESP电荷计算了,RED已没有必要使用了。
这里把各种程序做一下总结,是笔者讲授的北京科音分子动力学与GROMACS培训班中的一页幻灯片