在VMD中绘制Gaussian计算的分子振动矢量的方法
在VMD中绘制Gaussian计算的分子振动矢量的方法
Method for plotting molecular vibrational vectors calculated by Gaussian in VMD
文/Sobereva@北京科音
First release: 2020-Sep-8 Last update: 2024-Feb-4
Gaussian用户观看freq任务产生的振动矢量一般都是通过GaussView看(虽然也有ChemCraft等其它一些程序也可以看)。然而,起码对于GaussView 6来说,GaussView显示振动矢量的一个很大不足是箭头太细,而且头部不够粗,导致有时候都看不清楚,放在文章里不够美观。另外,GaussView绘制分子结构的作图选项不够灵活,而且还收费。VMD是极其流行的化学体系可视化程序,免费、灵活、图像效果好,本文介绍如何通过笔者写的VMD作图脚本非常方便地绘制Gaussian的振动分析任务产生的振动矢量。VMD可以在http://www.ks.uiuc.edu/Research/vmd/免费下载。
在这里下载笔者编写的绘图脚本和示例文件:http://sobereva.com/attach/567/file.zip。此脚本至少对于目前撰文时的VMD正式版中最新的1.9.3、Gaussian 09和16是完全适用的。
这里以绘制多巴胺的振动矢量为例进行演示。把文件包里的dopamine.out放到VMD目录下,这是多巴胺的Gaussian的freq任务的输出文件。然后我们得把这个.out文件转化成一个VMD可以认的结构文件的格式,比如可以把此文件载入GaussView,然后另存为.pdb或.mol2文件。也可以下载Multiwfn(http://sobereva.com/multiwfn),启动Multiwfn后载入此文件,然后选主功能100的子功能2,通过相应选项导出为.pdb或.xyz文件。
把文件包里的drawarrow.tcl和GauNorm.tcl都放到VMD目录下,然后用文本编辑器打开GauNorm.tcl,把开头的set filename后面的文件名改为dopamine.out。之后启动VMD,把多巴胺的结构文件载入VMD,然后在文本窗口输入source GauNorm.tcl执行此脚本,此时振动矢量信息就被读入了,与此同时定义了名为norm的绘制振动矢量的命令。之后在VMD的文本窗口输入比如norm 4,就可以把4号振动模式通过箭头画出来。
norm后面还可以接第2个参数,用来设置箭头长度是正则矢量的几倍,数值越大箭头越长,默认是3。norm后面还可以接第3个参数,用来设置箭头的半径,默认为0.05。比如norm 5 6 0.07就代表用6倍长度、0.07的半径绘制第5个振动矢量。默认是用黄色绘制箭头,如果想用别的颜色,把GauNorm.tcl中的draw color后面的yellow改成其它颜色名,比如cyan。
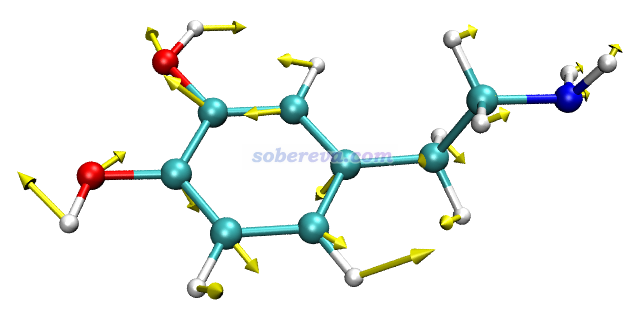
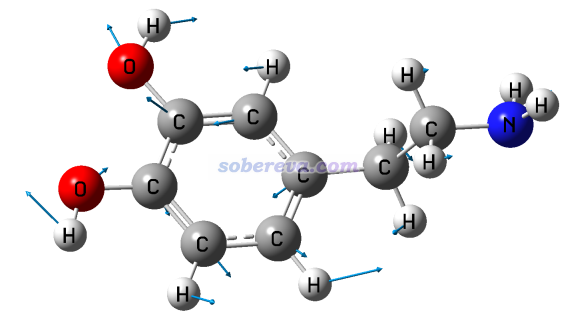
此例输入norm 17 5,然后令分子以CPK方式显示(在Graphics - Representation里把Drawing method改为CPK,再把Sphere Scale设为0.6),效果如下,可见非常理想!和GaussView显示的相对比,可见展现的信息是相同的,而GaussView画的箭头相比之下明显太小气了。
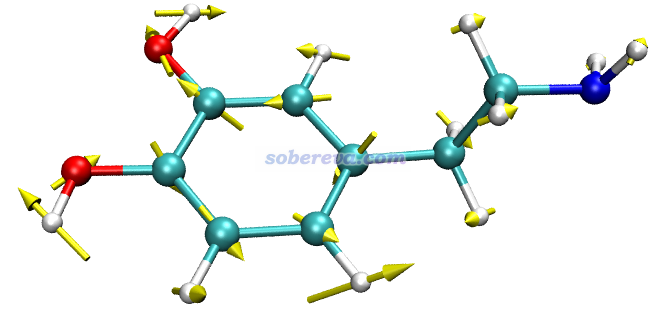
注意GauNorm.tcl开头还有个set ilinear语句,如果当前体系是线型体系,必须把后面的值改为1。
如果你在Gaussian做freq或opt freq任务中按照《在Gaussian中做限制性优化的方法》(http://sobereva.com/404)中的做法将N个原子的笛卡尔坐标冻结了,运行source GauNorm.tcl之前必须把里面set nfreeze后面的值设为N(默认为0,没有原子被冻结)。
如果你希望让箭头的始端位于各个原子上(和GaussView的风格一致),就把本文文件包里的drawarrow2.tcl放到VMD目录下,把GauNorm.tcl里的两处drawarrow都改为drawarrow2并保存。之后再按上文绘图即可。用norm 17 4命令,效果如下