关于为什么Multiwfn算的出RESP电荷与Antechamber的有所差异
关于为什么Multiwfn算的出RESP电荷与Antechamber的有所差异
Regarding why the RESP charge calculated by Multiwfn is different from that of Antechamber
文/Sobereva@北京科音 2019-Sep-27
0 前言
RESP电荷的原理、细节以及计算在《RESP拟合静电势电荷的原理以及在Multiwfn中的计算》(http://sobereva.com/441)有详细说明,Multiwfn是计算RESP电荷最方便易用和灵活的程序。有Multiwfn用户发现,Multiwfn算出的某些分子的RESP电荷与antechamber的不同,在此文我说一下原因。使用Multiwfn算RESP电荷的用户不是很有必要看此文,Multiwfn给的结果肯定是合理的。但如果想了解一些细节的话也可以看看。
简单来说导致差异的原因主要有两点:
(1)拟合点的分布不同
(2)等价约束设置不同
下文涉及的体系的相关文件可以在这里下:http://sobereva.com/attach/516/file.rar。本文用的是2019-Sep-26更新的Multiwfn 3.7(dev)版。
1 拟合点的分布不同带来的差异
Multiwfn在计算RESP电荷的时候,对于3.6、3.7版默认用的是MK方式设置拟合点(以后版本有可能会改变,届时看Multiwfn的RESP模块界面上的选项提示便知默认的是什么),拟合点位置由Multiwfn自己的代码设置,静电势由Multiwfn自己的代码计算或者调用cubegen计算。Antechamber计算RESP电荷时是从Gaussian的pop=MK IOp(6/33=2) IOp(6/42=6)输出文件里直接读取MK拟合点的坐标和静电势。虽然MK原文虽然定义了拟合点的分布规则,即每个原子上在不同原子半径倍数上有几层拟合点,但在具体确定拟合点坐标上,不同程序的细节不同,比如在球面上的拟合点以什么算法生成排布?拟合点的密度是多少?在原子接缝的地方是否自动去除过密的拟合点?等等。由于Multiwfn设置的拟合点的位置和Gaussian在这方面有一些不同,导致Multiwfn和Antechamber算的RESP电荷会有些许差别,但这个因素造成的差异通常不大,也没法说哪个程序更对,因为都是合理的。注:前述IOp(6/33=2)是让Gaussian把拟合点的位置和静电势数值打印到输出文件里的选项,而IOp(6/42=6)用来将拟合点的密度设为高于默认值。在Multiwfn的RESP电荷计算界面里也可以直接设拟合点的密度。
下面我们拿乙醇这个体系来对比一下。这里笔者直接用gview画了一个乙醇,保存成了.mol2文件。然后用antechamber产生了对应的gjf文件,里面的关键词是#HF/6-31G* SCF=tight Test Pop=MK iop(6/33=2) iop(6/42=6),原本带着的opt我给删了,对于本文的讨论没意义。然后用Gaussian 09 E.01进行计算,得到的chk文件我转成了fch文件。
用Multiwfn载入fch文件,进入RESP计算界面,直接选标准两步式RESP电荷计算,结果如下
Center Charge
1(C ) -0.250749
2(H ) 0.064449
3(H ) 0.064449
4(H ) 0.064449
5(C ) 0.547451
6(H ) -0.091298
7(H ) -0.091298
8(O ) -0.714589
9(H ) 0.407135
在计算第二步的时候提示用了以下等价性约束
Constraint 1: 2(H ) 3(H ) 4(H )
Constraint 2: 6(H ) 7(H )
再用antechamber计算RESP电荷,会出现一批文件,其中有两个RESP计算模块输出的文件ANTECHAMBER_RESP1.OUT和ANTECHAMBER_RESP2.OUT。第一个就是第一步拟合的输出,第二个就是第二步拟合的输出,最终结果看第二个里面的下面的部分
Point Charges Before & After Optimization
no. At.no. q(init) q(opt) ivary d(rstr)/dq
1 6 -0.229693 -0.229799 0 0.003990
2 1 0.077688 0.059374 0 0.000000
3 1 0.077682 0.059374 2 0.000000
4 1 0.034340 0.059374 2 0.000000
5 6 0.464072 0.533889 0 0.001841
6 1 -0.057092 -0.086155 0 0.000000
7 1 -0.057098 -0.086155 6 0.000000
8 8 -0.720426 -0.720426 -99 0.001375
9 1 0.410526 0.410526 -99 0.000000
q(init)是第一步拟合之后产生的电荷,q(opt)是经过第二步拟合最终得到的RESP电荷。ivary是等价性约束关系,比如no.为3和4的原子的ivary是2,就代表2、3、4在拟合过程中保持等价,这和前面看到的Multiwfn自动用的约束完全一样。8和9号原子的ivary为-99,代表它们在第二步拟合过程中电荷被约束为保持不变。
Antechamber这里产生的RESP电荷和前面给的Multiwfn算出来的有轻微差异,但顶多也就差0.01几。如果我们想让Multiwfn算出的电荷与Antechamber的精确一样,那就在选择开始计算RESP电荷之前先选一下选项8,然后再选1开始标准两步式拟合,按屏幕提示的要求把Gaussian输出文件路径输进去,这时Multiwfn就和Antechamber一样从Gaussian输出文件里读取拟合点的位置和静电势了,输出信息如下,和Antechamber给出的完全相同(个别原子仍有10的负六次方的差异是收敛限设置、舍入误差等数值细节导致的,完全可忽略不计)
Center Charge
1(C ) -0.229798
2(H ) 0.059373
3(H ) 0.059373
4(H ) 0.059373
5(C ) 0.533887
6(H ) -0.086155
7(H ) -0.086155
8(O ) -0.720426
9(H ) 0.410526
2 等价约束设置不同带的差异
下面我们看苯甲醛这个例子。哪怕Multiwfn也直接从Gaussian输出文件里读取拟合点的信息,结果与Antechamber给出的仍有轻微差异,这是由于两个程序用的等价性约束设置不同所带来的。
我们先用Multiwfn基于Gaussian里的拟合点信息计算RESP电荷。启动Multiwfn,载入本文文件包里的C6H5CHO.fchk,进入RESP模块,先选择8,再选择1,再输入C6H5CHO.out的路径,得到的结果如下
Center Charge
1(C ) -0.115868
2(C ) -0.111841
3(C ) -0.114944
4(C ) -0.146385
5(H ) 0.137991
6(H ) 0.134593
7(H ) 0.153718
8(H ) 0.139930
9(C ) 0.391423
10(H ) 0.059972
11(C ) -0.223448
12(H ) 0.155781
13(C ) 0.034578
14(O ) -0.495500
Sum of charges: 0.000000
RMSE: 0.001373 RRMSE: 0.079595
从屏幕上的提示可见,这个体系的RESP电荷计算过程只做了第一步,因为按照RESP电荷定义的规则,这个体系本身不需要做第二步。在第一步计算的时候没有自动做任何等价性约束。
也用antechamber进行计算,从给出的ANTECHAMBER_RESP2.OUT中可以看到所有原子的ivary都为-99,即第二步相当于什么也没做,因此最终得到的RESP电荷来自于第一步。然后看ANTECHAMBER_RESP1.OUT,输出信息为
no. At.no. q(init) q(opt) ivary d(rstr)/dq
1 6 0.000000 -0.101223 0 0.003514
2 6 0.000000 -0.148518 0 0.002793
3 6 0.000000 -0.139285 0 0.002916
4 6 0.000000 -0.148518 2 0.002793
5 1 0.000000 0.136078 0 0.000000
6 1 0.000000 0.140390 0 0.000000
7 1 0.000000 0.147385 0 0.000000
8 1 0.000000 0.140390 6 0.000000
9 6 0.000000 0.404823 0 0.001199
10 1 0.000000 0.028701 0 0.000000
11 6 0.000000 -0.139285 3 0.002916
12 1 0.000000 0.147385 7 0.000000
13 6 0.000000 0.001412 0 0.004999
14 8 0.000000 -0.469734 0 0.001041
可见结果与Multiwfn产生的略有差异,特别是第11号原子。从ivary可见,第一步拟合的时候约束了4=2、8=6、11=3、12=7。此体系结构如下
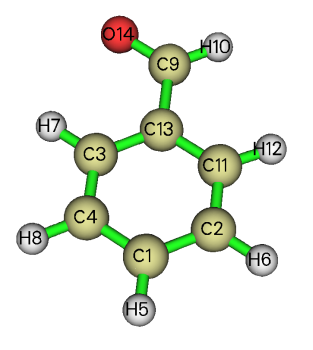
可见antechamber自动把苯环上左右对称的原子电荷约束为了相同,而Multiwfn默认情况下没有做这个约束,因此俩程序结果不同。
如果想让Multiwfn也实现同样的约束,自己写个约束设置文件即可。写个文本文件比如叫eqs.txt,内容为
4,2
8,6
11,3
12,7
在Multiwfn计算RESP电荷前,选择设置等价性约束的选项,再选读取约束文件,输入eqs.txt的路径,然后选择2开始一步式RESP电荷拟合,此时自定义的约束会被利用上,结果为
Center Charge
1(C ) -0.101222
2(C ) -0.148519
3(C ) -0.139284
4(C ) -0.148519
5(H ) 0.136078
6(H ) 0.140390
7(H ) 0.147385
8(H ) 0.140390
9(C ) 0.404824
10(H ) 0.028701
11(C ) -0.139284
12(H ) 0.147385
13(C ) 0.001411
14(O ) -0.469734
Sum of charges: 0.000000
RMSE: 0.002020 RRMSE: 0.117100
可见此时结果和Antechamber给出的精确相同了。
对苯环上对称的原子设置等价性约束并不是RESP电荷原文里提出的标准做法,纯粹是Amber的开发者自己定义的规则。这个规则有一点道理,但带来的坏处是会使得静电势重现性误差增加。如上述信息所示,不做这个约束时RMSE是0.001373,做约束后就增加到了0.002020。对于Multiwfn用户,这种约束想施加就施加,不想施加就不施加。如果懒得手动一条一条写这种等价性约束设置,在Multiwfn中可以直接利用结构的对称性自动产生等价性约束设置文件,看《RESP拟合静电势电荷的原理以及在Multiwfn中的计算》(http://sobereva.com/441)第3.5节的例子。