将GROMACS的原子电荷信息读入VMD的方法
2023-Jun-8注:推荐用http://bbs.keinsci.com/thread-37839-1-1.html里介绍的VMD载入tpr文件的插件,比本文的做法更方便。VMD载入tpr文件后直接就有了tpr中的原子电荷信息。
将GROMACS的原子电荷信息读入VMD的方法
Method to load GROMACS atomic charge information into VMD
文/Sobereva @北京科音
First release: 2017-Mar-27 Last update: 2022-Apr-12
《使VMD实时显示gromacs轨迹中原子的受力》(http://sobereva.com/36)
《使VMD读入Gromacs产生的trr轨迹中速度信息的方法》(http://sobereva.com/117)
有很多VMD里面的插件,比如显示偶极矩、计算静电势、绘制红外光谱的插件都依赖于原子电荷,有时候我们还需要编写依赖于原子电荷的脚本、使用依赖于原子电荷的选择语句,因此把GROMACS模拟时用的原子电荷载入VMD也是非常重要的,这里说说怎么做。
首先在这里下载笔者开发的gmxoutchg程序:http://sobereva.com/soft/gmxoutchg_1.1.rar
其中gmxoutchg.exe是Windows版可执行文件,没后缀的gmxoutchg是Linux下可执行文件,gmxoutchg.f90是源文件。经测试此程序兼容GROMACS 2016.1、2018.8和2019.3版,对其它版本兼容性未经测试。
tpr文件里蕴含了模拟所需的一切信息,包括原子电荷。用GROMACS里的dump命令将二进制的tpr文件转化成文本文件dump.txt,执行以下命令:
gmx dump -s test.tpr > dump.txt
然后将dump.txt放到gmxoutchg.exe所在目录下,运行gmxoutchg.exe,程序就会解析其中的数据,在当前目录下产生charge.txt。其中包含体系所有原子的原子电荷,顺序和结构文件里的原子顺序完全一致。其中第一列、第二列、第三列分别是原子电荷、分子序号、原子序号。
将charge.txt放到VMD目录后,在载入对应的结构/轨迹后,可以用以下tcl脚本将原子电荷从charge.txt中读入。
set sel [atomselect top all]
set natom [$sel num]
set rdchg [open "charge.txt" r]
set chglist {}
for {set iatm 0} {$iatm<=[expr $natom-1]} {incr iatm} {
gets $rdchg line
scan $line "%f" chg
lappend chglist $chg
}
$sel set charge $chglist
close $rdchg
如果想验证是否正确载入了,可以用[atomselect top all] get charge命令把所有原子的原子电荷输出出来(当然,也可以把all换成选择语句只输出指定部分的原子电荷)。
也可以将Coloring method设成Charge,直接从颜色上检验是否正确载入了。下面是乙醇盒子,在默认色彩刻度(RWB)下越红代表原子电荷越负,越蓝代表原子电荷越正。
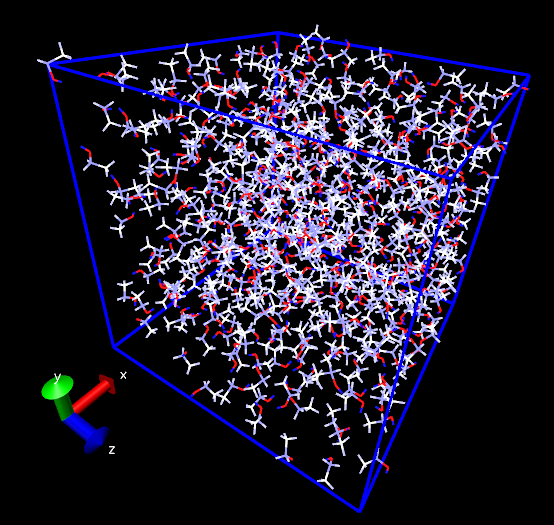
顺带一提,对于一些体系通过恰当设定显示方式,可以令原子电荷分布一目了然。比如下图的分子,一个显示方式设为Licorice并且把键调细,另一个显示方式是CPK,把圆球调大,用透明材质,用charge来着色,效果挺不错。
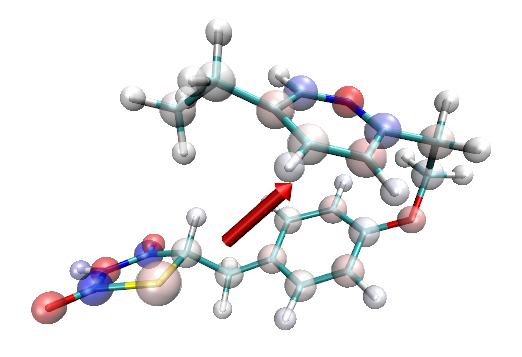
有了原子电荷信息可以直接使用VMD的依赖于原子电荷的插件了,比如Extensions-Visualization-Dipole Moment Watcher可以观看基于原子电荷计算的偶极矩矢量,就是上图的红色箭头。
PS:还有一种把原子电荷载入的方法是使用
gmx editconf -f nico.tpr -mead nico.pqr
这样nico.tpr里对应的原子电荷就被输出到了nico.pqr的电荷那一列了,pqr文件里还有原子半径信息,把nico.pqr载入VMD时也会读入其中的原子电荷。但这种做法致命缺点是原子电荷保留精度太低,只有两位小数,当涉及一些静电作用的分析时误差可能较大。