PM7、PM6-DH+半经验方法在优化碱基对儿时的失败
PM7、PM6-DH+半经验方法在优化碱基对儿时的失败
Failure of PM7 and PM6-DH+ semi-empirical methods in optimizing base pairs
文/Sobereva @北京科音
First release: 2013-Dec-30 Last update: 2015-Jul-18
已在《大体系弱相互作用计算的解决之道》(http://sobereva.com/214)当中介绍过PM7和PM6-DH+,这两种方法专门考虑了弱相互作用,对于优化弱相互作用大体系是很合适的。
笔者最近优化了一个GC碱基对儿体系,如下所示
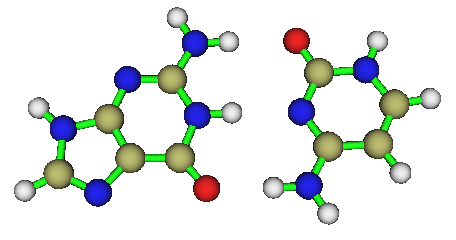
初始结构中这个碱基对儿是完全在同一个平面上的,真实情况也正是如此,然而用PM7优化后它们竟不在同一个平面上

用PM6-DH+优化也有这个问题。然而用更老的AM1、RM1、PM6,或PM6-DH2优化则都没有这个问题。至于PM3优化,结果则惨不忍睹!整体都弯了!虽然一般认为AM1和PM3半斤八两,对弱相互作用计算都很烂,不过对于这个体系的优化PM3可谓彻底失败,偏离平面不是一星半点,就连氨基都翘起来了而和单体自己不在同一个平面上。
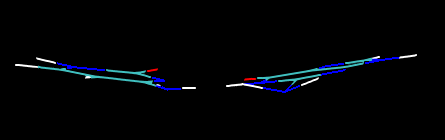
优化用的是MOPAC2012 Version 13.331W,且都加上了PRECISE关键词以让优化收敛得可以足够精确。
这个碱基对儿的优化结果表明,虽然PM7专门考虑了描述弱相互作用,但是终究只是半经验的水准,对于精细的研究,不要对其期望过高。用作大体系的优化或小体系预优化没问题,但是想算精确了,DFT-D水平的优化还是有必要的。笔者在前一段期间高精度研究氢气、氮气二聚体问题时也发现PM7不理想,它没能给出正确的二聚体构型稳定性顺序,而B3LYP-D3则基本和CCSD(T)的结论一致,见此贴《静电效应主导了氢气、氮气二聚体的构型》(http://sobereva.com/209)和对应的文章J. Mol. Model., 19, 5387-5395。
对于GC碱基对儿PM7优化后偏离平面的问题,笔者给PM7和MOPAC的作者J.J.P. Stewart写了邮件进行咨询,很快得到了答复
Dear Prof Lu,
I have checked the GC base pair - you are correct. This is a bug that I
did not know about. I agree that it is a bug.
Thank you for the information that this bug also shows up in PM6-DH+.
This is important, because PM7 uses a hydrogen bond function that is
similar to the one in "-DH+".
Just now work on adding "-D3H4" is being done in Prof Hobza's group. I
will inform them of your discovery.
For routine work, the error in "-DH+" looks small, but it is definite.
I'll discuss possible ways of fixing this fault with Hobza's group.
Best wishes to you for 2014.
Jimmy
看来果然不是程序的bug,而是PM7、PM6-DH+用的那种弱相互作用校正形式作崇。希望PM7的后继者能解决此问题,使之成为更可靠的处理弱相互作用体系的半经验方法。
2015-Jul-18补充:目前的MOPAC已经支持了PM6-D3H4和PM6-D3,笔者测试了一下,优化出来的体系是平面的,没有上述问题。而且根据DOI: 10.1021/acs.jctc.5b00296测试结果,PM7的精度明显明显不如PM6-D3H4,比PM6-D3差得更多,因此现在笔者不再推荐PM7,而推荐PM6-D3H4和PM6-D3。