调节平面分子间距的方法
调节平面分子间距的方法
The method to adjust the distance between planar molecules
文/Sobereva@北京科音 2013-Feb-3
计算化学研究中有时需要调节两个平面分子间距离,比如研究不同间距下的相互作用能。如果两个平面分子的坐标正好对着,或者分子平面平行于某个笛卡尔平面,那么改变间距很容易。前者就用gview的设定键长大小的工具就能调,后者就自行对笛卡尔坐标统一加减一定的数值就可以。比较麻烦的是分子既不平行于笛卡尔平面,两个分子也不是正对着,如下图这种情况
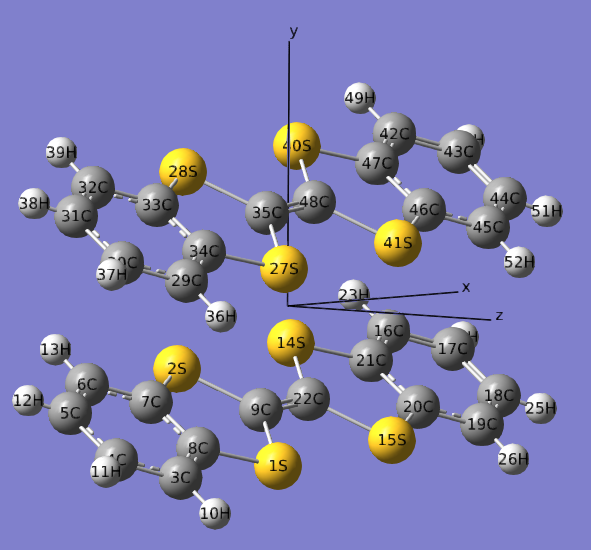
这里提供两个办法设定这两个分子平面的(垂直)间距。
方法1:
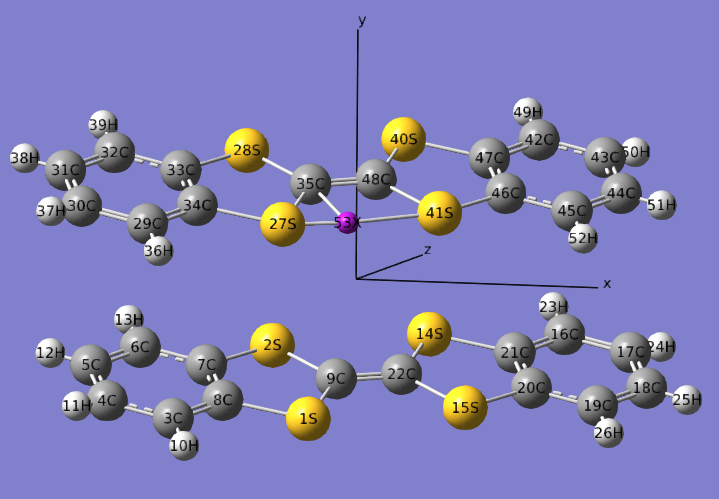
使用protoplane程序(/usr/uploads/file/20150610/20150610055901_52653.rar),得到一个分子上某原子在另一个分子平面上的(垂直)投影点坐标,并在此处设一个虚原子,然后用gview的键长设定工具调节这两个原子的间距(设定窗口中Atom1和Atom2都选为Translate group模式),两个分子就会相应地整体移动,从而调整了分子平面的间距。
protoplane程序启动后先依次输入三个原子的坐标来定义平面方程。然后再输入要投影的原子的坐标,就会输出投影后的坐标,以及此原子到平面的垂直距离。
输入的时候为了省事,可以直接把原子笛卡尔坐标从Gaussian输入文件里复制到DOS窗口内(先在文本编辑器里复制坐标,然后在DOS窗口标题栏上点右键,选编辑-粘贴)。之后,可以将生成的坐标直接拷贝到Gaussian输入文件里(DOS窗口标题栏上点右键,选编辑-复制,然后圈中要复制的区域,点回车,然后粘贴到文本编辑器里),并定义为X原子。
如下所示,将9号原子投影到27、35、41号原子定义的平面上就是53X,之后在gview里调节9C和53X的距离即可改变分子平面的间距了
protoplane程序源代码如下:
program prjtoplane
implicit real*8 (a-h,o-z)
write(*,*) "prjtoplane: Get the projected position of a point on a specific plane"
write(*,*) "Programmed by Sobereva (sobereva@sina.com), 2013-Feb-2"
write(*,*)
write(*,*) "You will input three point to define the plane"
write(*,*) "Input x,y,z coordinate of point 1 e.g. 2.3,1.0,-4.2 or 2.3 1.0 -4.2"
read(*,*) x1,y1,z1
write(*,*) "Input x,y,z coordinate of point 2"
read(*,*) x2,y2,z2
write(*,*) "Input x,y,z coordinate of point 3"
read(*,*) x3,y3,z3
do while(.true.)
write(*,*) "Input x,y,z coordinate the point you want to project"
read(*,*) x0,y0,z0
call pointprjple(x1,y1,z1,x2,y2,z2,x3,y3,z3,x0,y0,z0,prjx,prjy,prjz)
write(*,*)
write(*,*) "The x,y,z coordinate of the projected position is"
write(*,"(3f15.8)") prjx,prjy,prjz
write(*,"(a,f15.8)") " The vertical distance from the point to the plane is",dsqrt((prjx-x0)**2+(prjy-y0)**2+(prjz-z0)**2)
write(*,*)
write(*,*) "Now you can input another point you want to project, or press Ctrl+C to exit"
end do
end program
subroutine pointprjple(x1,y1,z1,x2,y2,z2,x3,y3,z3,x0,y0,z0,prjx,prjy,prjz)
real*8 x1,y1,z1,x2,y2,z2,x3,y3,z3,x0,y0,z0,prjx,prjy,prjz,A,B,C,D,t
call pointABCD(x1,y1,z1,x2,y2,z2,x3,y3,z3,A,B,C,D)
t=(D+A*x0+B*y0+C*z0)/(A**2+B**2+C**2)
prjx=x0-t*A
prjy=y0-t*B
prjz=z0-t*C
end subroutine
subroutine pointABCD(x1,y1,z1,x2,y2,z2,x3,y3,z3,A,B,C,D)
real*8 v1x,v1y,v1z,v2x,v2y,v2z,x1,y1,z1,x2,y2,z2,x3,y3,z3,A,B,C,D
v1x=x2-x1
v1y=y2-y1
v1z=z2-z1
v2x=x3-x1
v2y=y3-y1
v2z=z3-z1
A=v1y*v2z-v1z*v2y
B=-(v1x*v2z-v1z*v2x)
C=v1x*v2y-v1y*v2x
D=A*(-x1)+B*(-y1)+C*(-z1)
end subroutine
方法2:
这个方法是先让指定的分子平面平行于XY平面,然后自行对分子的Z坐标统一进行适当加减,就改变了分子平面间距。
2021-Aug-16注:下文用VMD的做法已经不推荐使用了!因为使用Multiwfn来实现方便得多,见《Multiwfn中非常实用的几何操作和坐标变换功能介绍》(http://sobereva.com/610)。让体系的一个平面平行于某笛卡尔平面的做法在此文2.4节专门有示例。在此文介绍的Multiwfn的子功能300的子功能7里用选项1还可以方便地让体系根据特定矢量进行平移。
这里提供一个VMD程序的Tcl脚本用于实现此目的。先将以下内容复制到VMD的命令行窗口执行
proc alignplane {ind1 ind2 ind3} {
set atm1 [atomselect top "serial $ind1"]
set atm2 [atomselect top "serial $ind2"]
set atm3 [atomselect top "serial $ind3"]
set vec1x [expr [$atm2 get x] - [$atm1 get x]]
set vec1y [expr [$atm2 get y] - [$atm1 get y]]
set vec1z [expr [$atm2 get z] - [$atm1 get z]]
set vec2x [expr [$atm3 get x] - [$atm1 get x]]
set vec2y [expr [$atm3 get y] - [$atm1 get y]]
set vec2z [expr [$atm3 get z] - [$atm1 get z]]
set sel [atomselect top all]
$sel move [transvecinv [veccross "$vec1x $vec1y $vec1z" "$vec2x $vec2y $vec2z"]]
$sel move [transaxis y 90]
}
然后在命令行窗口运行诸如alignplane 27 35 41就可以让27、35、41号原子定义的平面平行于XY平面,如下所示

现在可以利用file-save coordinates将当前结构保存出来,然后用诸如Excel/Origin之类或自写程序来平移分子。但是在VMD里也可以很方便地整体平移分子。
VMD会将自动将每个分子以fragment x描述。诸如前例,就有fragment 0和fragment 1代表这两个分子。假设要平移的是fragment 1,就先执行
set sel [atomselect top "fragment 1"]
然后比如向Z轴正方向平移0.5埃,即平移向量是0 0 0.5,就运行$sel move [transoffset {0 0 0.5}]