Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 Re: Quantum Chemistry » Large gap between two methods used to measure S0-T1 excitation energy » 2026-07-07 23:23:38
Thank you for the reply.
Indeed I found out that the E_ST gaps in gas-phase and solution-phase (external iteration) are:
delta-SCF method: 4.39 eV (gas-phase) / 4.44 eV (solution-phase / external iteration)
TDDFT method: 3.69 eV (gas-phase) / 3.70 eV (solution-phase / external iteration)
So the gas-phase difference seems to be the dominant factor. Would you then say that the delta-SCF method is probably more reliable?
Also I have two additional questions if you don't mind:
1. For this specific purpose of the delta-SCF method for vertical E_ST gap, am I correct that external iteration is meaningful, unlike for normal SCF?
2. Can I do the state-specific external iteration method for TDDFT calculation in ORCA? I can't find anything similar that is mentioned in the manual or tutorial.
If the answer is no, would you say that ORCA is fundamentally less powerful than G16 for calculation of excited states accompanying significant electronic structure reorganization, CT states for example, in highly polar solvents?
#2 Quantum Chemistry » Large gap between two methods used to measure S0-T1 excitation energy » 2026-07-06 07:14:15
- wham09
- Replies: 3
Dear Prof. Lu,
I am studying a S0 -> T1 excitation of an organic bimolecular encounter complex.
I initially used the following method:
1. S0 state calculation and save non-equilibrium solvation
%chk=S0.chk
# wB97XD/def2TZVP scrf=(read, solvent=dimethylsulfoxide)
0 1
[geometry]
NonEq=write
2. T1 state calculation with external iteration
%oldchk=S0.chk
%chk=T1.chk
# wB97XD/def2TZVP scrf=(externaliteration, read, solvent=dimethylsulfoxide) geom=check guess=read TD(nstates=5, triplets)
0 1
NonEq=read
Then I obtained the energy difference between #1 and #2.
Later, I tested the following method. Using the S0.chk from above:
%oldchk=S0.chk
%chk=T1-DFT.chk
# wB97XD/def2TZVP scrf=(externaliteration, read, solvent=dimethylsulfoxide) geom=check guess=read
0 3
NonEq=read
Then I obtained the energy difference between #1 and this calculation.
From the former method using S0 (SCF) vs. T1 (TD, external iteration), I got 335.2 nm wavelength, while from the latter method using S0 (SCF) vs. T1 (SCF, external iteration), I got 279.4 nm.
I checked the electronic structures of the two T1 states and they were similar (both the same valence-excited state). However, the excitation energy difference is quite large.
Which method would you say is more reliable? I assumed that for this case, it is okay to use external iteration for SCF also because the spin state is set different.
#3 Re: Quantum Chemistry » Problem of non-equilibrium solvation in SF-TDA » 2026-05-28 01:37:49
Dear Prof. Lu,
Again, thank you for the swift reply.
Your answer concerns me though, because if I'm correct, the general reliability of SF-TDA for solution-phase reactions becomes questionable.
Could you maybe recommend a route to communicate about this topic and find solutions? I do know that a lot of ORCA discussions are ongoing in Keinsci forum, but I'm not confident that an English thread (or a poorly translated Chinese thread) will be taken seriously there...
#4 Quantum Chemistry » Problem of non-equilibrium solvation in SF-TDA » 2026-05-28 00:12:59
- wham09
- Replies: 3
Dear Prof. Lu,
I have tested an organic molecule (neutral, singlet) in ORCA's spin-flip (SF-TDA) calculation.
Below is the general input command block that I used:
! BHANDHLYP def2-TZVP tightSCF CPCM(DMSO)
%cpcm xfeps 0.5 end
%TDDFT
SF TRUE
NROOTS 10
END
* xyz 0 3
...
As a result, the spin-flip ground-state was the singlet (as expected), and the spin-flip first excited-state shows S^2 of roughly 1.1, which seems to indicate the singlet diradical species.
My question is the following: When I do SF-TDA, the initial reference state is triplet, not singlet. Then, does the "slow" portion of solvation (that is fixed for non-equilibrium solvation calculation of spin-flip states) correspond to the triplet? If this is true, wouldn't this be unphysical, because in reality the slow portion of solvation should be that of the singlet ground-state?
#5 Re: Quantum Chemistry » Basis functions for 6-31G* used to fit SMD » 2026-03-19 18:58:18
Thank you very much.
I have one follow-up question about 6-31g*: When I tested the automatic density fitting of Gaussian 16 (I used PBEPBE/genecp/auto 6D 10F, with 6-31g* for main group elements and LANL2DZ for TM), the converged SCF energy differed significantly from the result of not using /auto. Depending on the structure, the difference reached over 30 kcal/mol. Moreover, a multi-step reaction energy profile changed drastically by using or not using /auto for SP calculations of the reactants and the products.
Is this because Gaussian’s density fitting is generally really bad? Or, is the automatically generated auxiliary basis set for PBE/6-31g* (or LANL2DZ) normally ill-defined? Or maybe the automatic density fitting is somehow sensitive to the type of d and f basis functions?
#6 Re: Quantum Chemistry » Basis functions for 6-31G* used to fit SMD » 2026-03-18 11:40:36
Additional question: For single-point energy calculation using an ordinary functional, if I want to compare the performance of the following Pople-type basis sets (6-31g*, 6-311g**, 6-311+g**) with def2-series (which use 5D 7F), would you say that it is more proper to use 6D 10F uniformly for all Pople-type basis sets?
#7 Quantum Chemistry » Basis functions for 6-31G* used to fit SMD » 2026-03-18 09:54:38
- wham09
- Replies: 5
Dear Prof. Lu,
I have so far evaluated SMD dissolution free energies of organometallic compounds using M05-2X/genecp (mixed 6-31G*/SDD), which should default to 5D/7F basis functions.
However, I realized that when M05-2X/6-31G* was used to fit the SMD model, the basis functions may have been 6D/10F (am I right?).
Should I actually specify 6D 10F when I use genecp to obtain accurate SMD energies? Or will the difference be negligible? I am currently testing it, but I would like to know if the basis function types actually matter for this type of energy calculation, based on a theoretical reasoning.
#8 Quantum Chemistry » Advantages of (meta-)GGA functionals » 2026-03-15 17:09:13
- wham09
- Replies: 1
Dear Prof. Lu,
If I understood your blog post correctly (http://sobereva.com/272), (meta-)GGA functionals are generally recommended when there is a risk of static correlation in ground state calculation, while not really recommended for anything else. (Please correct me if I'm wrong)
But I also know that nowadays, range-separated functionals such as wB97M-V are very sophisticated. And if I think about the concept of range-separation, I don't really see how a good-enough range-separated functional should be any worse than (meta-)GGA functionals even for moderate static correlation. And if static correlation was really severe, DFT will not be suitable anyways.
I understand the merits of B97-3c and r2SCAN-3c, but my question is: Does a traditional (meta-)GGA functional like PBE or TPSS have any reason to be utilized for non-periodic, non-TM-cluster calculations if I only care about accuracy?
#9 Re: Quantum Chemistry » Basis set requirement for anions under very high level of theory » 2026-03-15 16:31:48
I appreciate your replies!
#10 Re: Quantum Chemistry » Basis set requirement for anions under very high level of theory » 2026-03-13 00:23:50
Thank you for the reply. To further clarify, if the conditions you explained (significantly negative charge on certain atoms, and their participation in the reaction) are met, addition of diffuse functions to those atoms is required even if the basis set is already QZ or 5Z quality?
Additional: I notice that in the Multiwfn’s orca input generating function, the option for DLPNO-CCSD(T)/CBS (3,4/def2) disappears when I activate the diffuse function option, and only the option for extrapol(3,4/aug-cc) remains. Is this because ma-TZVPP/ma-QZVPP are somewhat less appropriate for extrapolation?
#11 Quantum Chemistry » Basis set requirement for anions under very high level of theory » 2026-03-12 17:27:09
- wham09
- Replies: 4
Dear Prof. Lu,
I wonder if diffuse functions are "always" required for reaction energy calculation of anions, even if the calculation level is already very high. For example, in the following 4 scenarios, should diffuse functions be added?
1. ordinary hybrid functional (e.g. PBE0-D4 or wB97M-V) with def2-QZVPP: Should change to ma-QZVPP?
2. double hybrid functional (e.g. PWPB95-D4 or wB97M(2)) with def2-QZVPP: Should change to ma-QZVPP?
3. DLPNO-CCSD(T) with CBS (def2-TZVPP / def2-QZVPP): Should change to ma-TZVPP -> ma-QZVPP?
4. DLPNO-CCSD(T) with CBS (cc-pVTZ / cc-pVQZ): Should change to aug-cc-pVTZ -> aug-cc-pVQZ?
#12 Quantum Chemistry » Judgement on DFT vs MR method selection » 2026-03-04 10:21:41
- wham09
- Replies: 1
Dear Prof. Lu,
I'm studying spin-delocalized states. Specifically, I'm looking at a transition state of radical transfer (A-B + C_dot -> A_dot + B-C), 3-center-3-electron bonding situation.
For this, I have the following questions:
1. Is it correct that this situation inherently requires multireference methods, and normal DFT is absolutely not suitable?
2. Is it reasonable (or common) to optimize the geometries of the reactants and the TS by DFT, obtain thermal correction and dissolution free energies by DFT, evaluate the single point gas-phase energies by MRPT, and then sum everything to obtain free energies?
3. Do the answers to 1 and 2 change if the radical donor "A" is 3rd-row (prone to degeneracy) or 4/5th-row (less prone to degeneracy, but relativistic effect emerges) transition metal?
4. Do the answers change if what I want to study is transition state of excited-state homolysis of carbon-carbon, carbon-heteroatom, or main group atom - transition metal bond (as a part of a much bigger molecule)?
Thank you very much.
#13 Quantum Chemistry » Applying Marcus theory by QM calculation » 2026-03-04 10:08:49
- wham09
- Replies: 0
Dear Prof. Lu,
I would like to apply semi-classical Marcus model to evaluate solution-state electron transfer rate constants, for which I have multiple questions.
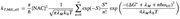
1. Let's say the ET reaction is A + B -> A(+) + B(-). In principle, should I calculate two sets of driving forces (delta_G), Huang-Rhys factors (S), the corresponding frequencies (omega), and reorganization energies (lambda) from separate calculations of A, A(+), B, B(-), and then combine them somehow?
Or should I optimize the geometry of A...B bimolecular complex, and then optimize A(+)...B(-) complex by TD optimization (following the CT state) and finally calculate one set of the factors?
2. In your blog post (sobereva.com/330), you stated that lambda_outer is difficult to calculate and often approximated. But in another post (sobereva.com/333), you presented quite clearly how to calculate it. Is the method presented in the article 333 indeed the standard way? Or is the accuracy of this approach questionable?
3. From your lecture notes and blog posts, I think I have some understanding of how to calculate all the terms required in the semi-classical Marcus equation, except the electronic coupling (NAC or H_ab). Is it correct that the most rigorous way to get this is to optimize the MECP geometry of the A…B complex, at which the intermolecular electron transfer actually happens, and then obtain the eigenvalue difference?
If there is other way that is more standard or commonly used, can it be done using Gaussian or ORCA?
4. If I wanted to calculate rate of outer-sphere electron transfer between an organic reactant and an electrode (in a solution of electrolyte in organic solvent), I could imagine that there will be many more uncertainties, such as:
1) how to optimize the distance between the electrode and the reactant at the moment of ET, and how to simulate the double layer environment at this distance
2) how to optimize geometry of the reactant in the presence of electric field, organic solvent, and electrolyte
3) how to calculate the terms in the Marcus equation when electrode is involved
About this question, I'm basically a novice. Can all these be addressed by QM methods (desirably using Gaussian or ORCA)? If not, is there a less rigorous, but good enough QM routine that researchers use?
#14 Re: Multiwfn and wavefunction analysis » w-tuning for n-th excited state » 2026-02-10 04:24:40
About the second question, could you please clarify a little more? I understand that the “change” of energy and electronic structure from the ground state is only affected by the polar part of solvation model. But, isn’t the reference electronic structure of the ground state still affected by the nonpolar part of solvation? For example, the electron density curve of fluoride anion is different when PCM or SMD model is used, although not much. If the ground state electronic structure is better represented by SMD model, shouldn’t this be consistently applied for the corresponding excited state?
#15 Re: Multiwfn and wavefunction analysis » w-tuning for n-th excited state » 2026-02-09 07:31:37
Dear Prof. Lu,
I have two questions about TD calculation, and since one of them is closely related to this thread, I’ll leave these here.
1. When the excited states of interest are local excitation states, is w-tuned LRC-functional still more ideal than PBE0?
2. If I wanted to look at electronic structure of a geometry-optimized excited state in solution (by extracting natural orbitals from density=current TD calculation), should using SMD model yield more realistic electronic structure than PCM, at least in principle?
#16 Re: Quantum Chemistry » Solvent participation after transition state » 2026-02-02 02:04:03
Thank you for the answer.
To explain my purpose more specifically, I attached another figure here, with a subsequent chemical step and an additional pathway.
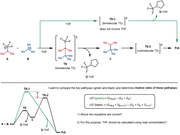
Basically, my confusion is coming from the fact that the reactants and the final product are exactly same in the two pathways, but the different "timing" of the THF complexation (to BF3) in the one-step pathway and the two-step pathway.
Could you please further clarify the additional two questions that are included in the figure?
#17 Quantum Chemistry » Solvent participation after transition state » 2026-01-31 09:09:34
- wham09
- Replies: 3
Dear Prof. Lu,
I'd like to ask for your advice about how to calculate the free energy change of a solution-phase reaction when solvent coordination seems to occur after the transition state.
For convenience of understanding what I mean, I attached the reaction diagram, explanations, and questions as a figure.

My questions are all in the figure, but I will also paste them below.
4. What would be the correct way here?
4-1) THF coordination to D happens after this reaction. dG = (G_C + G_D) – (G_A + G_B)
4-2) THF coordination to D happens during product formation. dG = (G_C + G_D-THF) – (G_A + G_B + G_THF)
5. If 4-2 is correct, should G_THF be calculated using the concentration as 12.3 M (neat solvent molarity) rather than 1.0 M? (when plugging into the dG equation)
Thank you very much in advance.
#18 Quantum Chemistry » Adjusting halogen radius for geometry optimization » 2026-01-31 03:24:00
- wham09
- Replies: 1
Dear Prof. Lu,
I am calculating a toluene-solvated organometallic transition state that contain half-cleaved palladium-bromide bond. I was optimizing the geometry of this TS using PBE0-D3(BJ)/ma-SVP (PCM, toluene) level of theory, and observed oscillating behavior.
Out of curiosity, I modified the PCM radius of bromine atom to 2.60 by using modifysph command, which is what was done in Truhlar's SMD18 paper (https://doi.org/10.1002/chem.201803652).
Of course, what Truhlar did was optimizing the geometries of his compounds using M06-2X/def2-TZVP (def2-TZVPD for bromine). I knew this, but I still chose to optimize my compound using PBE0-D3(BJ)/ma-SVP (PCM, toluene) and only change the PCM radius of bromine.
By doing this, the TS optimization was successful without oscillating behavior. Do you think that this could actually be a generally better way to calculate bromine-containing molecules, even when using functionals and basis sets that are different from the original SMD18 paper?
#19 Re: Multiwfn and wavefunction analysis » w-tuning for n-th excited state » 2026-01-30 00:33:07
Thank you for the answer.
I’d like to ask just one follow-up question.
The ground state of my molecule is doublet D0, and the first excited state (D1) is characterized by alpha-HOMO (30A) to alpha-LUMO (31A), while beta-HOMO stays 29B.
In this case, when I calculate N, N+1, and N-1 states for w-tuning, should I set the N+1 state as triplet? In this way the N+1 state will have the 31A populated.
#20 Multiwfn and wavefunction analysis » w-tuning for n-th excited state » 2026-01-28 02:42:36
- wham09
- Replies: 7
Dear Prof. Lu,
I am currently trying to conduct electron excitation analysis for the 5th excited state of my molecule.
The 5th excitation is characterized by charge transfer from alpha-HOMO to alpha-LUMO+3.
If I wanted to do w-tuning of long-range-corrected functional for this state, should I then look at N and N-1 (for HOMO), and separately, N+7 and N+6 (for LUMO+3)?
#21 Re: Quantum Chemistry » Questions on how to rigorously measure free energies in solution » 2026-01-20 05:49:12
I would be really grateful if you could explain the answer to Q1 a little more. The way I understand is that the G in solvent phase is as below:
G_sol = G_gas + dG_solv + 1.89
Because G_gas = E_gas + dG_corr_gas,
G_sol = (E_gas + dG_corr_gas) + dG_solv + 1.89
Here, dG_corr_gas should be the correction for gas-phase G. Then, shouldn’t I actually put E_gas into Shermo to obtain G_gas first, and then manually add dG_solv and 1.89kcal/mol?
#22 Quantum Chemistry » Questions on how to rigorously measure free energies in solution » 2026-01-20 03:23:59
- wham09
- Replies: 3
Dear Prof. Lu,
I did the following calculations for a ground-state molecule:
calc1. Geometry optimization (PCM model) + frequency calculation (B3LYP-D3(BJ) / def2-SVP, PCM)
calc2. Single point calculation (gas-phase) using calc1 geometry (B3LYP-D3(BJ) / def2-TZVP)
calc3. Single point calculation (gas-phase) using calc1 geometry (M05-2X / 6-31G*)
calc4. Single point calculation (SMD model) using calc1 geometry (M05-2X / 6-31G*, SMD)
From calc2, I obtained E_gas.
From calc3 and calc4, I obtained dG_solv.
If I understood correctly, the free energy is calculated as:
G = G_gas + dG_solv + 1.89
= (E_gas + dG_corr_gas) + dG_solv + 1.89
So I utilized Shermo on calc1 output for ZPVE scaling and msRRHO treatment, and obtained dG_corr_gas.
But here are my questions:
1. Am I correct to put E_gas (from calc2) into the Shermo's E value in the setting.ini file, rather than E_gas + dG_solv?
2. For calc3 and calc4 (using M05-2X functional), should superfine grids be used, as they sometimes should be for M06-2X?
3. In line with Q3, if I choose to do calc3 and calc4 using M06-2X instead, should I use superfine grids consistently?
4. calc1 was done with PCM because the geometry was sensitive to solvation. But then, is the Shermo result using the calc1 output truly the dG_corr_gas?
5. As an alternative, I did another frequency calculation (calc5) in the gas-phase using the calc1 geometry, but when I did this, I observed imaginary frequencies (expectedly). Would using Shermo on the calc5 output be more appropriate than using the calc1 output anyways?
6. Is there a truly ideal solution to Q4 and Q5? I don't care if it is cumbersome, I just want to know.
Thank you very much again for your time.
#23 Re: Multiwfn and wavefunction analysis » CT excited state energy and wavefunction analysis » 2026-01-15 05:22:59
About question 1: This is actually what I am most confused, because I thought I was correct based on your blog posts. Could you please correct me if I’m interpreting your texts in a wrong manner? (or wrong translation?) See below:
In your blog post “On the diffuse function and the "month" basis set (http://sobereva.com/119)”,
1. When is a diffusion function needed?
…
• Adding dispersion is very important and strongly recommended:
Calculate the weak interaction energy (if using a 4-zeta level basis set and not involving anions, then dispersion is not necessary)
…
• Adding dispersion can improve results, and should be done
optimize the structure of anionic or weakly interacting systems;
...
Also in “On the choice of basis sets in quantum chemistry (http://sobereva.com/336)”,
2. Selection of different task basis sets
2.2 Weak Interactions
…
The greater the proportion of dispersive interactions, the more necessary a diffuse function is required, and the more important the BSSE problem needs to be considered.
Basis set recommendations for weak interaction energy calculations:
Minimum: 6-31+G** or ma-def 2-SVP for DFT
…
Many thanks in advance!
#24 Re: Multiwfn and wavefunction analysis » CT excited state energy and wavefunction analysis » 2026-01-14 04:37:42
I’d be really grateful if you could provide some additional feedbacks, basically whether you agree to the following statements.
1. The structure (of which I’m studying the CT state) is a bimolecular interacting complex, which features intermolecular pi-stacking between two aromatic groups, and CT occurs between these two rings. In this case, even without significantly negatively charged atoms, it is recommended to add diffuse functions.
2. In the case of 1. or in case there are actually highly negatively charged atoms, I just have to accept that the method I have to use to get accurate energy (using diffuse functions) is not going to permit analysis of correct LUMO or LUMO+1.
Thank you very much.
#25 Multiwfn and wavefunction analysis » visualizing exchange and correlation holes in sobEDA framework » 2026-01-13 19:07:07
- wham09
- Replies: 1
Dear Prof. Lu,
I am wondering if I could somehow monitor the electron density change over the course of sobEDA steps so that I could separately examine exchange and correlation effects. Changes beginning from the promolecular state should be trivial, I would just have to substract e-density function between promol/frozen and frozen/final.
But ideally, I also want to dissect the first step (fragments to promolecular) into multiple different electronic structures to separate electrostatic, exchange, correlation, and dispersion effects on the electron density. What I could think of is repeating the fragment-to-promolecular step 1) with HF level, and 2) with the original level but without DFT-D3. (Original level I used for the whole sobEDA was PBE0-D3(BJ)/ma-TZVP).
Please let me know if this whole thing is just a stupid idea, or if it might be possible in a more sophisticated manner. Thank you very much!
#26 Re: Multiwfn and wavefunction analysis » CT excited state energy and wavefunction analysis » 2026-01-09 02:36:36
Thank you for the reply,
If you don’t mind, could you help me understand this in a little more detail? Because I thought that in a charge-transfer state where certain atoms (that received charge) are significantly negatively charged, at least those atoms should need to be treated with diffuse functions.
#27 Multiwfn and wavefunction analysis » CT excited state energy and wavefunction analysis » 2026-01-08 03:11:33
- wham09
- Replies: 7
Dear Prof Lu,
I have a neutral doublet structure (D0) whose first excited state (D1) turns out to be a charge-transfer state, which shows charge-separation (significantly negative charges on specific atoms and positive charges on other atoms). From what I learned, I would need to add diffuse functions to accurately measure the excitation energy.
At the same time, I want to study the D1 state's electronic state, examine molecular orbitals, etc. But I noticed that with diffuse functions used, the alpha-HOMO in the D1 state (alpha-LUMO in the D0 state) is severely broken in shape. Only when I do not add diffuse functions I see excitation to the correctly shaped orbital.
How should I resolve both energy evaluation and accurate wavefunction analysis at the same time?
#28 Re: Multiwfn and wavefunction analysis » Miscellaneous questions on TDDFT/electron-hole analysis » 2025-11-09 08:46:46
question 8: From what I understood your comments, in the figure that I attached (green: non-excited PES, orange: excited PES), is it correct that calculating accurate relative Gibbs free energies of all 6 points under the same calculation level is not feasible?
Especially because, for my specific molecule, the CT state of interest is assigned as the 2nd excited state when calculated with wB97XD, but is correctly (I think) assigned as the 1st excited state only when the w-tuned LC-wPBE is used.
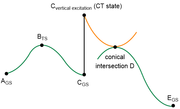
question 9: Also related to question 8, if I optimize the MECI structure with CASSCF, is it possible to calculate its delta G relative to other states on the reaction path calculated by DFT methods?
Additional question about Q9: As I explained above, the CT state of my molecule is assigned differently depending on the functional. So I'm not confident that CASSCF will assign the CT state as the 1st excited state. Then, should I increase the number of electrons and orbitals in the active space to be safe?
Additional question (new): I would think that state-specific solvation of the vertical excitation state will affect vibrational frequencies differently (in principle) from the linear response. Can I do excited state freq calculation under this exact state-specific environment? Perhaps do TD=read from the state-specific solvated TD calculation chk?
#29 Re: Multiwfn and wavefunction analysis » Miscellaneous questions on TDDFT/electron-hole analysis » 2025-11-08 13:03:43
About question 1: To be more specific, after TD optimization is complete, and then if I want to run single-point calculation of the excited state to obtain total Gibbs free energy (following the procedure you suggested for the question 7), would you recommend DFT-D3(BJ) for the functionals I mentioned?
About question 5: Just like question 1, for single-point calculation of the optimized excited state, SMD is preferred, right?
About question 8: I guess the question was not specifically about CAM-B3LYP. For example, I could use LC-wPBE with w-tuning specific to the structure of B*. In this case, I thought that the specifically tuned LC-wPBE is probably not adequate for calculation of other ground states and transition states in the whole reaction path (which could be 10~20 steps), compared to more general functionals such as PBE0-D3(BJ) or B3LYP-D3(BJ). Is it okay to use a more general functional for non-excited states and compare their free energies with the free energy of B* calculated by tuned LC-wPBE?
About question 9: I do have knowledge on conical intersections, although not an expert. My guess is perform relaxed scans (from the structure of B* to C) with 1) standard and 2) TD settings, and find out a scan point where the two energies are most similar? Could you let me know if there is a better, or a more appropriate, procedure?
#30 Multiwfn and wavefunction analysis » Miscellaneous questions on TDDFT/electron-hole analysis » 2025-11-07 03:48:11
- wham09
- Replies: 5
Dear Prof. Lu,
I hope to get help with several questions on TDDFT calculation for electron-hole analysis.
1. For TDDFT, whether the functional is PBE0, LC-wPBE, CAM-B3LYP, or wB97XD, would you generally recommend DFT-D3(BJ)?
2. Is LC-wHPBE really superior to LC-wPBE as Gaussian advertises?
3. Can the same w-tuning procedure be applied whether the ground-state is singlet, doublet, or triplet, and whether it is neutral, anionic, or cationic?
4. Can electron excitation and a bond-formation or bond-cleavage reaction be concerted? Can it be calculated?
5. Is it more recommended (or not recommended) to do TD calculations with SMD rather than PCM?
6. For excited state optimization, can I just use nosymm keyword and skip the manual arbitrary breaking of symmetry?
7. For calculation of total Gibbs energy of an optimized excited state, please check if what I describe below seems correct:
1) ES optimization (with GS checkpoint file available):
wB97XD/def2-SVP TD(Nstates=5, Root=2) Opt SCRF=solvent=dimethylsulfoxide geom=modify ... geometry modification
2) Take the structure and .chk file from 1). Run freq calculation:
wB97XD/def2-SVP TD(read, Nstates=5, Root=2) Freq SCRF=solvent=dimethylsulfoxide guess=read
3) Take the structure and .chk file from 1). Run SP calculation (gas-phase):
wB97XD/def2-TZVP TD(Nstates=5, Root=2) guess=read
4) Take the structure and .chk file from 1). Run SP calculation (gas-phase):
M052X/6-31G* TD(Nstates=5, Root=2) guess=read
5) Take the structure and .chk file from 1). Run SP calculation (solution-phase):
M052X/6-31G* TD(Nstates=5, Root=2) SCRF=solvent=dimethylsulfoxide guess=read
6) Calculate E = E(step 3) + E(step 5) - E(step 4). Put it into Shermo and use .out file from step 2. Get thermal correction.
8. Suppose I have a reaction pathway A -> TS-1 -> B -> B* -> TS-2 -> C. The excited state B* is a charge-transfer state, for which I would want to use a long-range-corrected functional, such as CAM-B3LYP. But CAM-B3LYP would be less appropriate for calculation of other states A, B, TS-1, and TS-2. But then, for reaction coordinate energy calculations, shouldn't the functional be consistently used for every molecule from A to C?
9. For the TS-2 between B* and C in the question 8, am I looking for "excited state of transition state"? I don't know how to think about it.