Acetaldehyde Excitation and Emission Cycle
This keyword requests that a calculation be performed in the presence of a solvent by placing the solute in a cavity within the solvent reaction field.
The Polarizable Continuum Model (PCM) using the integral equation formalism variant (IEFPCM) is the default SCRF method. This method creates the solute cavity via a set of overlapping spheres. It was initially devised by Tomasi and coworkers and Pascual-Ahuir and coworkers [Miertus81, Miertus82, Pascual-Ahuir94, Cossi96, Barone97, Cances97, Mennucci97, Mennucci97a, Barone98, Cossi98, Barone98a, Cammi99, Cossi99, Tomasi99, Cammi00, Cossi00, Cossi01, Cossi01a, Cossi02, Cossi03]. This model corresponds to SCRF=PCM. See [Tomasi05] for a recent review. The model of Chipman [Chipman00] is closely related to this method [Cances01].
Other available models are IPCM, which uses a static isodensity surface for the cavity [Foresman96], the Self-Consistent Isodensity PCM (SCIPCM) model [Foresman96], and the Onsager model [Kirkwood34, Onsager36, Wong91, Wong91a, Wong92, Wong92a], which places the solute in a spherical cavity within the solvent reaction field.
Many aspects of the PCM implementation have changed from Gaussian 03. In Gaussian 09, we use a continuous surface charge formalism that ensures continuity, smoothness and robustness of the reaction field, which also has continuous derivatives with respect to atomic positions and external perturbing fields [Scalmani10]. This is achieved by expanding the apparent surface charge that builds up at the solute-solvent interface in terms of spherical Gaussian functions located at each surface element in which the cavity surface is discretized. Discontinuities in the surface derivatives are removed by effectively smoothing the regions where the spheres intersect.
This formalism, initially proposed in 1999 by Karplus and York for the conductor screening model [York99], never received the attention it deserved. In Gaussian 09, we develop and generalize it within the framework of the PCM family of solvation methods, and it is now the default method for building the solute’s cavity and computing the reaction field.
The PCM method in Gaussian 09 includes an external iteration procedure whereby the program computes the energy in solution by making the solvent reaction field self-consistent with the solute electrostatic potential (the latter being generated from the computed electron density with the specified model chemistry) [Improta06, Improta07]. The difference with the standard approach (based on the variational approach or linear response theory) can be illustrated with MP2. The default procedure computes the solvent effect on the SCF density and then applies MP2 perturbation, while the external iteration approach computes the solvent effect self-consistently with respect to the MP2 density. While this technique is of primary interest for studying excited state processes such fluorescence, it can also be used for ground state calculations with theoretical methods that provide gradients: e.g. post-SCF methods. Use the ExternalIteration option to specify this method.
There are two basic approaches available for modeling excited states in solution:
Computing the lowest excited states in the solvent environment. This approach, which was also available in Gaussian 03, adds SCRF to a normal excited state calculation such as TD or CIS. This technique employs a linear response formalism by adding the necessary terms to the excited state method equations (thereby including the solvent effects on the excited states) [Cammi00, Cossi01]. In Gaussian 09, the geometry of a specific excited state can be optimized in solution with CIS or TD [Scalmani06].
A single excited state can be modeled via a state-specific approach. In this case, the program computes the energy in solution by making the electrostatic potential generated by the excited state density self consistent with the solvent reaction field [Improta06, Improta07], using the external iteration technique mentioned above.
For excited state calculations in solution, there is a distinction between equilibrium and non-equilibrium calculations. The solvent responds in two different ways to changes in the state of the solute: it polarizes its electron distribution, which is a very rapid process, and the solvent molecules reorient themselves (e.g., by a rotation), a much slower process. An equilibrium calculation describes a situation where the solvent had time to fully respond to the solute (in both ways), e.g., a geometry optimization (a process that takes place on the same time scale as molecular motion in the solvent). A non-equilibrium calculation is appropriate for processes which are too rapid for the solvent to have time to fully respond, e.g. a vertical electronic excitation.
Equilibrium solvation is the default for CIS and TD-DFT excited state geometry optimizations. Non-equilibrium is the default for CIS and TD-DFT energies using the default PCM procedure, and equilibrium is the default for calculations using the external iteration approach (SCRF=ExternalIteration). See the examples for the method for computing non-equilibrium external iteration calculations.
By default, CASSCF PCM [Cossi99] calculations correspond to equilibrium calculations with respect to the solvent reaction field/solute electronic density polarization process. Calculation of non equilibrium solute-solvent interaction involving two different electronic states (e.g. the initial and final states of a vertical transition) can be performed using the NonEq=type PCM keyword, in two separate job steps (see the PCM input section below).
Keywords and options specifying details for a PCM calculation (i.e., the default SCRF=PCM or SCRF=CPCM) may be specified in an additional blank-line terminated input section provided that the Read option is also specified. Keywords within this section follow general Gaussian input rules. The available keywords are listed in a separate subsection following the examples.
For the Onsager model (SCRF=Dipole), the solute radius in Angstroms and the dielectric constant of the solvent are read as two free-format real numbers on one line from the input stream. A suitable solute radius is computed by a gas-phase molecular volume calculation (in a separate job step); see the discussion of the Volume keyword.
For the IPCM and SCIPCM models, the input consists of a line specifying the dielectric constant of the solvent and an optional isodensity value (the default for the latter is 0.0004).
Solvent=item
Selects the solvent in which the calculation is to be performed. Note that the solvent may also be specified in the input stream in various ways for the different SCRF methods. If unspecified, the solvent defaults to water. Item is a solvent name chosen from the list at the end of this section.
PCM
Performs a reaction field calculation using the integral equation formalism model (IEFPCM). This is the default. Some details of the formalism and the implementation have changed with respect to Gaussian 03, as described in [Scalmani10]. IEFPCM is a synonym for PCM.
When PCM is used for an anisotropic or ionic solvent, then items in the PCM input section must be used to select the anisotropic and ionic dielectric models for these types of solvents, using the Read option (see below). The continuous surface charge formalism is also not available with such solvents, and no derivatives can be computed.
CPCM
Performs a PCM calculation using the CPCM polarizable conductor calculation model [Barone98, Cossi03].
Dipole
Performs an Onsager model reaction field calculation.
IPCM
Performs an IPCM model reaction field calculation. Isodensity is a synonym for IPCM.
SCIPCM
Performs an SCIPCM model reaction field calculation, i.e. the SCRF calculation uses a cavity determined self-consistently from an isodensity surface.
Read
Indicates that a separate section of keywords and options providing calculation parameters should be read from the input stream (as described above). This must be specified for anisotropic and ionic solvents.
Checkpoint
Retrieves the SCRF information from the checkpoint file.
Modify
Retrieves the SCRF information from the checkpoint file, and also reads modifications from the input stream.
ONIOMPCM=k
Performs an ONIOM calculation in solution [Vreven01, Mo03] according to the scheme selected by the code letter k, for which these are the valid values:
| A | The reaction field is computed self-consistently using the integrated ONIOM density (available only for energies, and not available for semi-empirical methods). | |
| B | The reaction field is computed for the real-system at the low-level and the corresponding polarization charges are used as external charges in the sub-calculations on the model-systems (available only for energies). | |
| C | The reaction field is computed only for the real-system at the low-level while the sub-calculations on the model-systems are performed assuming zero reaction field (i.e., gas phase). This selection is available for energies, optimizations and frequencies. | |
| X | The reaction field is computed separately in each sub-calculation always using the cavity of the real-system. This is the default if ONIOM and SCRF are specified (available for energies, optimizations and frequencies). |
ExternalIteration
Does a self-consistent PCM calculation performing an external iteration through Link 124. This approach computes the energy in solution by making the solute’s electrostatic potential self-consistent with the solvent reaction field [Improta06, Improta07] (discussed above). ExternalIteration is available only for energy calculations. SelfConsistent and SC and StateSpecificPerturbation and SSPerturbation are synonyms for this option.
Restart
Restarts a PCM external iteration calculation from the checkpoint file.
SkipVacuum
Performs the first iteration in an external iteration PCM calculation in solution. This is useful to accelerate the convergence toward self-consistency, and it is the default. We recommend using this option to obtain correct excited state ordering in a TD-DFT calculation. DoVacuum performs the first iteration in the gas phase; this option is required in order to compute a ΔG of solvation.
SMD
Do an IEFPCM calculation with radii and non-electrostatic terms for Truhlar and coworkers’ SMD solvation model [Marenich09]. This is the recommended choice for computing ΔG of solvation, which accomplished by performing gas phase and SCRF=SMD calculations for the system of interest and taking the difference the resulting energies.
G03Defaults
Modify the PCM defaults in order to reproduce the results of a Gaussian 03 PCM calculation as closely as possible. Note that perfect agreement is not always possible due to improvements in the program.
A0=val
Sets the value for the solute radius a0 in the route section (rather than reading it from the input stream of an SCRF=Dipole calculation). If this option is included, then Solvent or Dielectric must also be included.
Dielectric=val
Sets the value for the dielectric constant of the solvent. This option overrides Solvent if both are specified.
GradVne
Use Vne basins for the numerical integration.
GradRho
Use density basins for the numerical integration. The job may fail if non-nuclear attractors are present.
UseDensity
Force the use of the density matrix in evaluating the density.
UseMOs
Force the use of MOs in evaluating the density.
GasCavity
Use the gas phase isodensity surface to define the cavity rather than solving for the surface self-consistently. This is mainly a debugging option.
COSMORS
Produces the data file used by COSMO/RS and other programs.
The following table details the availability of the various SCRF=PCM calculation types by theoretical method:
| Method | Energy (Ext. Iter.) | Energy | Opt | Freq | 3rd Order Props a | NMR |
| MM | no | yes | yes | yes | no | no |
| AM1, PM3, PM3MM, PM6, PDDG | no | yes | yes | yes | no | no |
| HF, DFT | yes | yes | yes | yes | yes | yes |
| MP2 | yes | yesb | yesb | yesb | yesb,c | yesb |
| MP3, MP4(SDQ), CCSD, QCISD, BD | yes | yesb | no | no | no | no |
| CASSCF | yes | yes | yes | yese | no | no |
| CIS | yes | yesd | yesd | yesd | no | no |
| TD | yes | yesd | yesd | yesd,e | no | no |
| ZIndo | no | yes | no | no | no | no |
aFor example, Freq=Raman, ROA or VCD; bComputed via SCF MO polarization; cRaman intensities are computed numerically (i.e., as with Freq=NRaman); dUsing the linear response approach; eNumerical frequencies only.
CIS and CASSCF frequencies with PCM solvation must be done numerically using Freq=Numer.
Restarting SCRF Calculations. SCRF=ExternalIteration and SCRF=IPCM jobs can be restarted from the read-write file by using the Restart option. SCRF=SCIPCM calculations that fail during the SCF iterations should be restarted via the SCF=Restart keyword.
Non-Default Methods. The IPCM model is available for HF, DFT, MP2, MP3, MP4(SDQ), QCISD, CCD, CCSD, CID, and CISD energies only. The SCIPCM model is available for HF and DFT energies and optimizations and numerical frequencies. The Onsager (SCRF=Dipole) model is available for HF, DFT, MP2, MP3, MP4(SDQ), QCISD, CCD, CCSD, CID, and CISD energies, and for HF and DFT optimizations and frequency calculations. However, the Opt Freq keyword combination may not be used in SCRF=Dipole calculations.
PCM Energy. In general, energy output from the default SCRF method appears in the normal way within the output file. For example, here are the sections of the output file containing the predicted energy from a Hartree-Fock and from an MP2 PCM calculation:
Hartree-Fock SCRF calculation:
SCF Done: E(RHF) = -99.4687828290 A.U. after 8 cycles
Convg = 0.2586D-08 -V/T = 2.0015
MP2 SCRF calculation:
E2 = -0.1192799427D+00 EUMP2 = -0.99584491345297D+02
The predicted energy in solution includes all computed corrections (unlike in Gaussian 03 output).
Additional output lines may appear when various PCM options are included. For example, the following output is produced by an HF SCRF(SMD) calculation:
SCF Done: E(RHF) = -99.4687828290 A.U. after 8 cycles
Convg = 0.2586D-08 -V/T = 2.0015
SMD-CDS (non-electrostatic) energy (kcal/mol) = 0.54
(included in total energy above)
For state-specific and external iteration SCRF calculations, the final energy is computed by Link 124, which controls the external iteration, and is reported in a separate output section, which will appear very near the end of the output file, as in the following example:
--------------------------------------------------------------------
Self-consistent PCM results
===========================
<psi(f)| H |psi(f)> (a.u.) = -99.577537 (A)
<psi(f)|H+V(f)/2|psi(f)> (a.u.) = -99.584002 (B)
(Polarized solute)-Solvent (kcal/mol) = -4.06 (C)
--------------------------------------------------------------------
Partition over spheres:
Sphere on Atom Surface Charge GEl GCav GDR
1 H1 15.27 -0.157 -2.36 0.00 0.00
2 F2 32.58 0.157 -1.70 0.00 0.00
--------------------------------------------------------------------
Predicted energy value for external iteration and state-specific SRCF calculations
After PCM corrections, the energy is -99.5840023899 a.u.
--------------------------------------------------------------------
Line (A) reports the energy computed using the polarized solute wavefunction and the gas phase Hamiltonian, line (B) reports energy computed using the polarized solute wavefunction and the Hamiltonian in solution, line (C) reports the interaction energy between the polarized solute and the solvent, which corresponds to the integral <Ψ(f)|V(f)/2|Ψ(f)> (in kcal/mol), and the final line reports the predicted energy incorporating all PCM corrections.
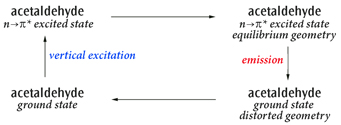
Fluorescence example: Emission (Fluorescence) from First Excited State (n→π*) of Acetaldehyde
Here we study the cycle:
Acetaldehyde Excitation and Emission Cycle
The primary process of interest is the emission, but this example shows how to study the complete cycle including the solvent effects.
Step 1: Ground state geometry optimization and frequencies (equilibrium solvation). This is a standard Opt Freq calculation on the ground state including PCM equilibrium solvation.
%chk=01-ac # B3LYP/6-31+G(d,p) Opt Freq SCRF=(Solvent=Ethanol) Acetaldehyde ground state 0 1 C C,1,RA X,2,1.,1,A O,2,RB,3,A,1,180.,0 X,1,1.,2,90.,3,0.,0 H,1,R1,2,A1,5,0.,0 H,1,R23,2,A23,5,B23,0 H,1,R23,2,A23,5,-B23,0 H,2,R4,1,A4,3,180.,0 RA=1.53643 RB=1.21718 R1=1.08516 R23=1.08688 R4=1.10433 A=62.1511 A1=110.51212 A23=109.88119 A4=114.26114 B23=120.56468
Here is the energy of the ground state optimized geometry in solution:
SCF Done: E(RB3LYP) = -153.851761719 A.U. after 1 cycles
Step 2: Vertical excitation with linear response solvation. This is a TD-DFT calculation of the vertical excitation, therefore at the ground state equilibrium geometry, with the default solvation: linear response, non-equilibrium. We perform a single-point TD-DFT calculation, which defaults to non-equilibrium solvation. The results of this job will be used to identify which state or states are of interest and their ordering. These results give a reasonable description of the solvation of the excited state, but not quite as good as that from a state-specific solvation calculation. In this case, we see that the n→π* state is the first excited state. Next, we will use the state-specific method to produce a better description of the vertical excitation step.
%oldchk=01-ac %chk=02-ac # B3LYP/6-31+G(d,p) TD=NStates=6 SCRF=(Solvent=Ethanol) Geom=Check Guess=Read Acetaldehyde: linear response vertical excited states 0 1
The vertical excitation (absorption) to first excited state from the non-equilibrium solvation linear response calculation:
Excited State 1: Singlet-A" 4.3767 eV 283.28 nm f=0.0000 <S**2>=0.000
Thus, the ground state to first excited state absorption is at 283.28 nm, computed via the linear-response approach.
Step 3: State-specific solvation of the vertical excitation. This will require two job steps: first the ground state calculation is done, specifying NonEq=write in the PCM input section, in order to store the information about non-equilibrium solvation based on the ground state. Second, the actual state-specific calculation is done, reading in the necessary information for non-equilibrium solvation using NonEq=read, and specifying the checkpoint file from Step 1:
%oldchk=01-ac %chk=03-ac # B3LYP/6-31+G(d,p) SCRF=(Solvent=Ethanol,Read) Geom=Check Guess=Read Acetaldehyde: prepare for state-specific non-eq solvation by saving the solvent reaction field from the ground state 0 1 NonEq=write --link1-- %chk=03-ac # B3LYP/6-31+G(d,p) TD(NStates=6,Root=1) SCRF=(Solvent=Ethanol,ExternalIteration,Read) Geom=Check Guess=Read Acetaldehyde: read non-eq solvation from ground state and compute energy of the first excited with the state-specific method 0 1 NonEq=read
Here is the energy of first excited state—at the ground state optimized geometry—from the non-equilibrium solvation state-specific calculation:
After PCM corrections, the energy is -153.687679826 a.u.
Subtracting this energy from the ground state energy (from step 1) gives the ground state to first excited state absorption including the state-specific solvation correction: at 277.69 nm.
Step 4: Relaxation of the excited state geometry. Next, we perform a TD-DFT geometry optimization, with equilibrium, linear response solvation, in order to find the minimum energy point on the excited state potential energy surface. Since this is a TD-DFT optimization, the program defaults to equilibrium solvation. As is typical of such cases, the molecule has a plane of symmetry in the ground state but the symmetry is broken in the excited state, so the ground state geometry is perturbed slightly to break symmetry at the start of the optimization. We retrieve the geometry and other data from the checkpoint file from Step 2:
%oldchk=02-ac %chk=04-ac # B3LYP/6-31+G(d,p) TD=(Read,NStates=6,Root=1) SCRF=(Solvent=Ethanol) Geom=Modify Guess=Read Opt=ReadFC Acetaldehyde: excited state opt Modify geometry to break Cs symmetry since first excited state is A" 0 1 4 1 2 3 10.0 5 1 2 7 -50.0
Here are the results for the first excited state after the geometry optimization of first excited state in solution (equilibrium geometry):
Excited State 1: Singlet-A 3.2074 eV 386.55 nm f=0.0013 <S**2>=0.000 12 -> 13 0.70615 This state for optimization and/or second-order correction. Total Energy, E(TD-HF/TD-KS) = -153.705918726
Step 5: Vibrational frequencies of the excited state structure. Now we run a frequency calculation to verify that the geometry located in step 4 is a minimum. The results could also be used as part of a Franck-Condon calculation if desired (see below). This is a numerical frequency calculation.
%oldchk=04-ac %chk=05-ac # B3LYP/6-31+G(d,p) TD=(Read,NStates=6,Root=1) Freq SCRF=(Solvent=Ethanol) Geom=Check Guess=Read Acetaldehyde excited state freq 0 1
The frequency calculation is used to confirm that the geometry optimized in Step 4 is a minimum on the excited state potential energy surface.
Step 6: Emission state-specific solvation (part 1). This step does state-specific equilibrium solvation of the excited state at its equilibrium geometry, writing out the solvation data for the next step via the PCM NonEq=write input.
%oldchk=05-ac %chk=06-ac # B3LYP/6-31+G(d,p) TD=(Read,NStates=6,Root=1) SCRF=(Solvent=Ethanol,ExternalIteration,Read) Geom=Check Guess=Read Acetaldehyde emission state-specific solvation at first excited state optimized geometry 0 1 NonEq=write
Here is the energy of first excited state—at its optimized geometry—from the equilibrium solvation state-specific calculation:
After PCM corrections, the energy is -153.707148980 a.u.
Step 7: Emission to final ground state (part 2). Finally, we compute the ground state energy with non-equibrium solvation, at the excited state geometry and with the static solvation from the excited state.
%oldchk=06-ac %chk=07-ac # B3LYP/6-31+G(d,p) SCRF=(Solvent=Ethanol,Read) Geom=Check Guess=Read Acetaldehyde: ground state non-equilibrium at excited state geometry. 0 1 NonEq=read
Here is the energy of ground state from a non-equilibrium solvation calculation in solution, using the first excited state optimized geometry and the solvent reaction field in equilibrium with the first excited state density):
SCF Done: E(RB3LYP) = -153.822024722 A.U. after 10 cycles
The difference between the energies from steps 6 and 7 gives the vertical emission energy. In this case, the first excited state to ground state emission, including the state-specific solvation correction, is at 396.63 nm.
Steps 1, 2, and 4 would be sufficient to compute the excitation and emission energies in the gas-phase (along with step 5 to confirm the nature of stationary point). They are not sufficient when solvent effects are included because the energies computed in step 4 correspond to the ground state solvent reaction field, while the emission takes place in the reaction field created in response to the excited state charge distribution. This is what is accounted for properly in steps 6 and 7.
If the band shape is to be calculated, then in the gas phase one would simply run a calculation with Freq=(ReadFC,FC,Emission), giving the checkpoint file from step 1 as the main checkpoint file for the job, and providing the name of the checkpoint file from step 5 in the input stream to specify the other state. For the solvated band shape, one must do Freq=(ReadFC,FC,Emission,ReadFCHT) using the checkpoint files for steps 1 and 5, but also providing the state-specific emission energy in the input section for the Franck-Condon calculation.
Additional input keywords may be specified for PCM SCRF calculations. They are placed in a separate input section, terminated as usual by a blank line, as in this example:
# B3LYP/6-31G(d) 5D SCRF(SMD,Solvent=Generic,Read) Water, solvation by methanol, re-defined as generic solvent. 0 1 O H,1,0.94 H,1,0.94,2,104.5 stoichiometry=C1H4O1 Input section for PCM keywords solventname=methanol eps=32.63 epsinf=1.758 … Blank line terminates PCM input
The PCM input section ends as usual with a blank line.
The following keywords are available for controlling PCM calculations (arranged in groups of related items).
The solvent for the PCM calculation is generally specified using the normal Solvent option to the SCRF keyword. You can use the following keywords to override some default values for known solvents.
Eps=x | Specifies the static (or zero-frequency) dielectric constant of the solvent. | |
EpsInf=x | Specifies the dynamic (or optical) dielectric constant of the solvent. | |
RSolv=x | Specifies the solvent radius (in Angstroms). Relevant only when using AddSph or Surface=SAS. |
Unspecified parameters default to the values for the solvent specified with the Solvent option (or to water if this option is omitted).
NonEq=item | Compute and save the non-equilibrium reaction field after the completion of an HF, DFT or CASSCF calculation using SCRF(Read), or at the end of any SCRF(ExternalIteration,Read) calculation. NonEq=Write says to save the data in the checkpoint file. Use NonEq=Read to retrieve it from the checkpoint file in a subsequent calculation. | |
Dis | Computes and includes in the total energy the solute-solvent dispersion interaction energy using the model of Floris and Tomasi [Floris89, Floris91]. The default is NoDis. This option cannot be used in a SCRF=SMD calculation. | |
Rep | Includes the solute-solvent repulsion interaction energy in the total energy using the model of Floris and Tomasi [Floris89, Floris91]. The default is NoRep. This option cannot be used in a SCRF=SMD calculation. | |
Cav | Includes the solute cavitation energy in the total energy using the model of Pierotti [Pierotti76]. The default is NoCav. This option cannot be used in a SCRF=SMD calculation. | |
CavityFieldEffects | Includes the effects of the cavity-field interaction energy (also known as local field effect) in the total energy according to the model of Cammi and co-workers [Cammi00a]. The default is not to include this effect. | |
CF=Eps=x | Specifies a different value for the static dielectric constant to be used in the cavity-field energy contribution. This is useful to modulate the magnitude of the cavity-field effects. | |
CF=EpsInf=x | Specifies a different value for the dynamic (optical) dielectric constant to be used in the cavity-field energy contribution. This is useful to modulate the magnitude of the cavity-field effects. | |
FitPot | Performs analysis of the solute-solvent interaction energy in terms of atomic or atomic groups additive contributions. This analysis involves a fitting of atomic charges to the molecular electrostatic potential in solution. | |
Iterative | Solves the PCM electrostatic problem to calculate polarization charges through an iterative method. | |
MxIter=N | Specifies the maximum number of iterations allowed to the iterative solution of the electrostatic problem. 400 is the default. | |
QConv=type|N | Sets the convergence threshold for the iterative calculations of the PCM polarization charges to 10-N or to one of the following predefined types: VeryTight (10-12), Tight (10-9) and Sleazy (10-6). The default is QConv=Tight. | |
SC=QConv=x | Specifies the convergence for the PCM polarization charges during the external iteration procedure. | |
MaxExtIt=x | Specify the maximum number of iterations allowed during the external iteration procedure. |
Anisotropic | Performs a PCM calculation for an anisotropic solvent according to the IEFPCM formalism. The 3-rank symmetric tensor representing the dielectric constant must be specified as the values for these six additional keywords: EPSX, EPSY, EPSZ, EUPHI, EUTHE, and EUPSI (all of them take a parameter: e.g., EPSX=value). | |
Ionic | Performs a PCM calculation for ionic solution according to the IEFPCM formalism. The ionic strength in mol/dm3 has to be specified as the value to the keyword DISM. |
By default, the program builds up the cavity using the UFF radii, which places a sphere around each solute atom, with the radii scaled by a factor of 1.1. There are also three United Atom (UA) models available (see below).
The cavity can be extensively modified in the PCM input section: changing sphere parameters and the general cavity topology, adding extra spheres to the cavity built by default, and so on. The whole molecular cavity can be also provided by the user in the input section.
Radii=model | Indicates the topological model and/or the set of atomic radii used. Available models and sets are: | |
UFF: Uses radii from the UFF force field scaled by 1.1. Hydrogens have individual spheres (explicit hydrogens). This is the default. | ||
UA0: Uses the United Atom Topological Model applied on atomic radii of the UFF force field for heavy atoms. Hydrogens are enclosed in the sphere of the heavy atom to which they are bonded. This was the default in Gaussian 03. | ||
UAHF: Uses the United Atom Topological Model applied on radii optimized for the HF/6-31G(d) level of theory. | ||
UAKS: Uses the United Atom Topological Model applied on radii optimized for the PBE1PBE/6-31G(d) level of theory. | ||
Pauling: Uses the Pauling (actually Merz-Kollman) atomic radii (uses explicit hydrogens). | ||
Bondi: Uses the Bondi atomic radii (uses explicit hydrogens). | ||
PDens=x | Sets the average density of integration points on the surface, in units of Angstrom-2. 5.0 is the default. Increasing this value results in a finer surface discretization. | |
Alpha=scale | Specifies the electrostatic scaling factor by which the sphere radius is multiplied. The default value is 1.1. | |
Surface=type | Specify the type of molecular surface representing the solute-solvent boundary. Available options are: | |
VDW: Van der Waals surface. Uses atomic radii (scaled) and skips the generation of “added spheres” to smooth the surface. This is the default. | ||
SES: Solvent Excluding Surface. The surface is generated by the atomic or group spheres and by the spheres created automatically to smooth the surface (“added spheres”). This was the default in Gaussian 03. | ||
SAS: Solvent Accessible Surface. The radius of the solvent is added to the unscaled radii of atoms and/or atomic groups. | ||
ModifySph | Alters parameters for one or more spheres. The modified spheres can be indicated in the PCM input using the following format: | |
ModifySph | ||
where atom is the atom number or element type. A slash may replace the radius in which case the internal default value is used. This is useful when you want provide an alpha value but do not need to modify the default radius. | ||
ExtraSph=N | Adds N user-defined spheres to the cavity. Parameters of the spheres can be indicated using the following format: | |
ExtraSph=N | ||
where X,Y,Z are the Cartesian coordinates in the standard orientation. | ||
NSph=N | The cavity is built just from the N spheres provided by the user, specified on lines of the following format: | |
atom_number radius [alpha] | ||
Specifying spheres by atom number mimics standard cavity behavior, while specifying Cartesian produces a fixed cavity which does not move with the structure. | ||
where X,Y,Z are the Cartesian coordinates in the standard orientation. | ||
SphereOnH=N | When using a United Atom Topological model, places an individual sphere on the hydrogen at the Nth position in the atoms list. | |
SphereOnAcidicHydrogens | When using a United Atom Topological model, puts individual spheres on acidic hydrogens (those bonded to N, O, S, P, Cl and F atoms). | |
OFac=x | Specifies the overlap index between two interlocking spheres [Pascual-Ahuir94] for SES added spheres. Decreasing this index results in a smaller number of added spheres. The default value is 0.89. | |
RMin=x | Sets the minimum radius in Angstroms for SES added spheres. Increasing this value results in a smaller number of added spheres. The default value is 0.2. |
GeomView | Create the file points.off describing the cavity. This file contains input for the GeomView program (see www.geomview.org), which can be used to visualize the molecular cavity. |
The following solvent keywords are accepted with the SCRF=Solvent option. We list the ε values here for convenience, but be aware it is only one of many internal parameters used to define solvents. Thus, simply changing the ε value will not define a new solvent properly.