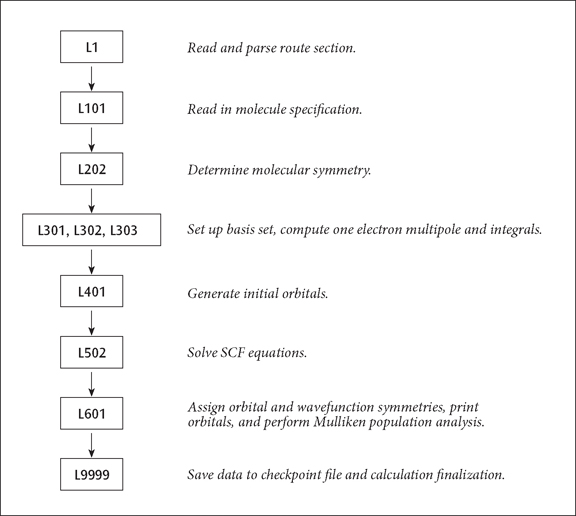
A Simple Route Sequence
This section documents keywords and options useful for developers who are extending and/or interfacing to Gaussian 09. It also discusses non-standard routes and the determination of the standard orientation.
The keywords and options described here are useful for developing new methods and other debugging purposes, but are not recommended for production level calculations.
Restart
We discuss here the general use of Restart, designed for debugging. See the Restart section for production use. This keyword restarts a calculation by reusing the read-write file. The form Restart L1 reuses the read-write file but generates a new route.
Restarts using the original route can specify the occurrence of a particular link and whether to clean up or retain overlay and link-volatile files using the following syntax:
#P Restart [Ln[(m)]] [Clean|KeepOverlay|KeepAll]
When all parameters are specified, the job restarts at the mth occurrence of Link n. Clean requests that all routine and overlay volatile files be removed by Link1, KeepOverlay requests that overlay-volatile files be retained but not link-volatile ones, and KeepAll retains everything. The default is to KeepAll if the read-write file is set up for an intra-link restart and Clean otherwise.
IOp1=keyword
This keyword controls various details of the operating system interface. The options are standard, but not all are implemented (or even relevant!) in every version.
OPTIONS
| FileIODump | Dump FileIO tables at the end of each link. FDump is a synonym for this keyword. | |
| TimeStamp | Turn on time stamping. TStamp is a synonym for this keyword. | |
| FileIOPrint | Turn on additional debug print in FileIO. | |
| Synch | Currently a no-op (appears in a few test jobs). | |
| NoDFTJ | Turn off use of the pure Coulomb term for non-hybrid DFT (seldom useful in production jobs). | |
| AbelianOnly | Force the use of only abelian symmetry (seldom useful in production jobs). | |
| NoPackSort | Turn off packing addresses into 32-bits during sorts, even if the address space being sorted is < 231 (seldom useful in production jobs). |
IOp2
This option sets the maximum amount of memory which will be dynamically allocated. MDV and Core are synonyms for IOp2.
IOp33
This sets the standard debug print option as specified.For example, the following sets IOp(33) to 3 in all invocations of overlay 2, and IOp(33) to 1 in all invocations of overlay 7:
IOp33(2=3,7=1)
The Gaussian 09 IOps Reference also documents all internal options (IOps). They are also documented on our web site: www.gaussian.com/iops.htm.
CPHF
The following options are used for debugging:
OPTIONS
| KeepMicro | Keep all EE centers in CPHF, even for Opt=CalcFC or Opt=CalcAll with non-quadratic microiterations, where atoms that are not used in internal coordinates need not be included in the CPHF. | |
| NoReuse | Do not reuse the electric field CPHF solution in the 2nd (nuclear) CPHF during frequency calculations. The default is ReUse. | |
| XY | Treat real and imaginary perturbations together. The opposite is NoXY, which does them separately. The default is to treat them separately if nuclear perturbations are also being done, but to treat them together if there are only electromagnetic perturbations. | |
| ZVector | Use the Z-Vector method [Diercksen81, Diercksen81a, Handy84] for post-SCF gradients. Allowed and the default if Hartree-Fock 2nd derivatives are not also requested. The NoZVector keyword says to use the full 3 × NAtoms CPHF for post-SCF gradients. |
FMM
The following options are available for debugging:
OPTIONS
| LMax=N | Specifies the maximum order multipole. The default is 25. | |
| Levels=N | Specifies the number of levels to use in the FMM. The default is 8 for molecules and is adjusted dynamically for PBC. | |
| Tolerance=N | Specifies the accuracy level as 10-N. The default values for N are 11 except for pass 0 of the SCF where it is 7. | |
| JBoxLen=N | Sets the minimum box length (size) to N/1000 Bohrs when doing J. By default, N is 2.5. The maximum of JBoxLen and KBoxLen is used if J and K are done at the same time. BoxLen is a synonym for JBoxLen. | |
| KBoxLen=N | Sets the minimum box length (size) to N/1000 Bohrs when doing K. By default, N is 0.75. The maximum of KBoxLen and JBoxLen is used if K and J are done at the same time. | |
| AllNearField | Turn on all near-field in FMM. | |
| NoParallelCPHF | Forbid parallel execution in FMM during the CPHF phase. NoParCPHF is a synonym for this option. |
Integral
The following options are used for debugging:
OPTIONS
| CNDO | Do calculation in main code using CNDO/2 integrals. | |
| INDO | Do calculation in main code using INDO/2 integrals. | |
| ZIndo1 | Do calculation in main code using ZIndo/1 integrals. | |
| ZIndoS | Do calculation in main code using ZIndo/S integrals. | |
| DPRISM | Use the PRISM algorithm [Gill94] for spdf integral derivatives. This is the default. | |
| Rys1E | Evaluate one-electron integrals using the Rys method [Dupuis76, King76, Rys83], instead of the default method. This is necessary on machines with very limited memory. | |
| Rys2E | If writing two-electron integrals, use Rys method (L314) [Dupuis76, King76, Rys83, Schlegel84]. This is slower than the default method, but may be needed for small memory machines and is chosen by default if regular (non-Raffenetti) integrals are requested (by the NoRaff option). | |
| DSRys | Use scalar Rys integral derivative code. Can combine with Berny for df only using Rys. | |
| Berny | Use Berny sp integral derivative and second derivative code (L702). | |
| Pass | Pass specifies that the integrals be stored in memory via disk, and NoPass disables this. Synonymous with SCF=[No]Pass, which is the recommended usage. | |
| NoJEngine | Forbid use of special Coulomb code. | |
| NoSP | Do not use the special sp integral program (L311) when writing integrals to disk. | |
| RevDagSam | Reverse choice of diagonal sampling in Prism. | |
| NoSchwartz | Do not use Schwartz integral estimates (only use the heuristic set). Schwartz says to use the Schwartz integral estimates in addition to the heuristic set. The default is to use both. | |
| RevRepFock | Reverse choice of Scat20 vs. replicated Fock matrices. | |
| NoDFTCut | Turn off extra DFT cutoffs. | |
| SplitSP | Split AO S=P shells into separate S and P shells. NoSplitSP is the default. | |
| SplitSPDF | Split AO S=P=D and S=P=D=F shells into S=P, D, and F. NoSplitSPDF is the default. | |
| SplitDBFSPDF | Split density S=P=D and S=P=D=F into S=P, D, and F. NoSplitDBFSPDF is the default. | |
| NoGather | Forbid use of gather/scatter digestion, even when processing small numbers of density matrices. Splatter is a synonym for this option. | |
| ForceNuc | Do nuclear-electron Coulomb with electron-electron. | |
| SepJK | Do J and K in HF/hybrid DFT separately for testing. | |
| Seq2E | Set up for parallel 2 electron integral evaluation but then do not run in parallel (for debugging). | |
| SeqXC | Set up for parallel 2 electron integral evaluation but then do not run in parallel (for debugging). | |
| SeqLinda | Cause Linda workers to run sequentially. Currently just makes the Linda workers other than the master run simultaneously but before the master. | |
| BigAtoms | Make all atom sizes large in XC quadrature. | |
| BigShells | Make all shell sizes large in XC quadrature. | |
| NoSymAtGrid | Do not use (Abelian) symmetry to reduce grid points on symmetry-unique atoms. | |
| LinMIO | Convert to linear storage in FoFCou for testing. | |
| RevDistanceMatrix | Reverse choice of whether to precompute distance matrix during numerical quadrature. The default is to precompute for molecules but not for PBC. | |
| NoDynParallel | Turn off dynamic work allocation. |
Sparse
The following options are used for debugging:
OPTIONS
| Loose | Sets the cutoff to 5 * 10-5. | |
| Medium | Sets the cutoff to 5 * 10-7. This is the default for semi-empirical methods. | |
| Tight | Sets the cutoff to 1 * 10-10. This is the default for DFT methods. | |
| N | Sets the cutoff to 1 * 10-N. |
ExtraLinks
This requests that additional links be executed. They are added to all instances of their overlay after the regular links. For example, ExtraLinks=L9997 will cause each instance of overlay 99 to include links 9999 (by default) and 9997, in that order.
ExtraOverlays
This command requests that extra overlay cards be read in non-standard route format and inserted into the standard route immediately before the final (overlay 99) card.
Skip
Skip initial overlay cards in the route. Skip=OvNNN skip until first occurrence of overlay NNN. Skip=M skip first M cards.
Use=Lnnn
This specifies alternate routes through the program.
OPTIONS
| L123 | Use L123 instead of L115 for IRC. This is the default for IRC, except for IRCMax jobs. | |
| L402 | Use old link 402 code for semi-empirical. | |
| L503 | Use link 503 for SCF. | |
| L506 | Use link 506 for ROHF. |
If a combination of options or links is required which is drastically different than a standard route, then a complete sequence of overlays and links with associated options can be read in. The job-type input section begins with the line:
# NonStd
This is followed by one line for each desired overlay, in execution order, giving the overlay number, a slash, the desired options, another slash, the list of links to be executed, and finally a semicolon:
Ov/Opt=val,Opt=val,…/Link,Link,…;
For example:
7/5=3,7=4/2,3,16;
specifies a run through the links 702, 703, and 716 (in this order), with option 5 set equal to 3 and option 7 equal to 4 in each of the links. If all options have their default value, the line would be
7//2,3,16;
A further feature of the route specification is the jump number. This is given in parentheses at the end of the link list, just before the semicolon. It indicates which overlay line is executed after completion of the current overlay. If it is omitted, the default value is +0, indicating that the program will proceed to the next line in the list (skipping no lines). If the jump number is set to -4, on the other hand, as in
7//2,3,16(-4);
then execution will continue with the overlay specified four route lines back (not counting the current line).
This feature permits loops to be built into the route and is useful for optimization runs. An argument to the program chaining routine can override the jump. This is used during geometry optimizations to loop over a sequence of overlay lines until the optimization has been completed, at which point the line following the end of the loop is executed.
Note that non-standard routes are not generally created from scratch but rather are built by printing out and modifying the sequence produced by the standard route most similar to that desired. This can be accomplished most easily with the testrt utility.
A Simple Route Example. The standard route:
# RHF/STO-3G
causes the following non-standard route to be generated:
1/38=1/1; 2/12=2,17=6,18=5,40=1/2; 3/6=3,11=1,16=1,25=1,30=1,116=1/1,2,3; 4//1; 5/5=2,38=5/2; 6/7=2,8=2,9=2,10=2,28=1/1; 99/5=1,9=1/99;
The resulting sequence of programs is illustrated below:
A Simple Route Sequence
The basic sequence of program execution is identical to that found in any ab initio program, except that Link 1 (reading and interpreting the route section) precedes the actual calculation, and that Link 9999 (writing to the checkpoint file) follows it. Similarly, an MP4 single point has integral transformation (links 801 and 804) and the MP calculation (link 913) inserted before the population analysis (Link 601) and Link 9999. Link 9999 automatically terminates the job step when it completes.
A Route Involving Loops. The standard route:
# RHF/STO-3G Opt
produces the following non-standard route:
1 1/18=20,19=15,38=1/1,3; 2 2/9=110,12=2,17=6,18=5,40=1/2; 3 3/6=3,11=1,16=1,25=1,30=1,71=1,116=1/1,2,3; 4 4//1; 5 5/5=2,38=5/2; 6 6/7=2,8=2,9=2,10=2,28=1/1; 7 7//1,2,3,16; 8 1/18=20,19=15/3(2); 9 2/9=110/2; 10 99//99; 11 2/9=110/2; 12 3/6=3,11=1,16=1,25=1,30=1,71=1,116=1/1,2,3; 13 4/5=5,16=3/1; 14 5/5=2,38=5/2; 15 7//1,2,3,16; 16 1/18=20,19=15/3(-5); 17 2/9=110/2; 18 6/7=2,8=2,9=2,10=2,19=2,28=1/1; 19 99/9=1/99;
The resulting sequence of program execution is illustrated below:
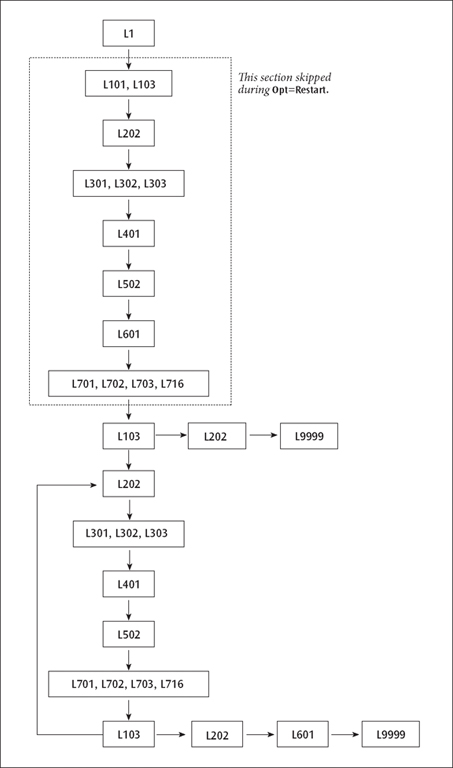
A Route Involving Loops
Several considerations complicate this route:
The first point of the optimization must be handled separately from later steps, since several actions must be performed only once. These include reading the initial molecule specification and generating the initial orbitals.
There must be a loop over geometries, with the optimization program (in this case the Berny optimizer, Link 103) deciding whether another geometry was required or the structure has been optimized.
If a converged geometry is supplied, the program should calculate the gradients once, recognize that the structure is optimized, and quit.
Population analysis and orbital printing are done by default only at the first and last points, not at the relatively uninteresting intermediate geometries.
The first point has been dealt with by having two basic sequences of integrals, guess, SCF, and integral derivatives in the route. The first sequence includes Link 101 (to read the initial geometry), Link 103 (which does its own initialization), and has options set to tell Link 401 to generate an initial guess. The second sequence uses geometries produced in Link 103 in the course of the optimization, and has options set to tell Link 401 to retrieve the wavefunction from the previous geometry as the initial guess for the next.
The forward jump on the eighth line has the effect that if Link 103 exits normally (without taking any special action), the following lines (invoking Links 202 and 9999) are skipped. Normally, in this second invocation of Link 103, the initial gradient will be examined and a new structure chosen. The next link to be executed will be Link 202, which processes the new geometry, followed by the rest of the second energy+gradient sequence, which constitutes the main optimization loop. If the second invocation of Link 103 finds that the geometry is converged, it exits with a flag which suppresses the jump, causing Links 202, 601 and 9999 to be invoked by the following lines and the job to complete.
Lines 11-16 form the main optimization loop. This evaluates the integrals, wavefunction, and gradient for the second and subsequent points in the optimization. It concludes with Link 103. If the geometry is still not converged, Link 103 chooses a new geometry and exits normally, causing the backward jump on line 16 to be executed, and the next line processed to be line 11, beginning a new cycle. If Link 103 finds that the geometry has converged, it exits and suppresses the jump, causing the concluding lines (17-19) to be processed.
The final instance of Link 601 prints the final multipole moments as well as the orbitals and population analysis if so requested. Finally, Link 9999 generates the archive entry and terminates the job step.
MP and CI optimizations have the transformation and correlation overlays (8 and 9) and the post-SCF gradient overlays (11 and 10, in that order) inserted before overlay 7. The same two-phase route structure is used for numerical differentiation to produce frequencies or polarizabilities.
The route for Opt=Restart is basically just the main loop from the original optimization, with the special lines for the first step omitted. The second invocation of Link 103 is kept and does the actual restarting.
Before a calculation is performed, a molecule can be reoriented to a different coordinate system, called the standard orientation, with the use of molecular symmetry. In geometry optimizations, reorientation occurs at every step; the program then checks if the standard orientation of a molecule has flipped by 180 degrees during an optimization and avoids the flip. This avoids jumps when animating optimizations, IRCs, etc. in GaussView and improves SCF convergence.
This section describes the goals, factors to consider, and various rules for positioning axes for the standard orientation of molecules.
The goals for selecting conventions for standard orientation are:
To simplify the 3x3 transformation matrices by reorienting the molecule.
Two Z-matrices differing in values of internal coordinates but identical in integer quantities (such as occurs on subsequent points of a geometry optimization) should produce the same standard orientation.
Two different Z-matrices for the same molecule should produce the same coordinates, except for a possible renumbering of atoms.
Maximizing the number of molecular orbital coefficients which are zero by symmetry.
The factors that should be considered for standard orientation are:
A right handed coordinate system is used throughout the calculation.
The molecule is translated so that its center of charge is at the origin.
Atoms are not reordered relative to their order upon input.
The Cartesian axes are considered to increase in priority in the order X < Y < Z.
Criteria for rotating and aligning an axis are listed below. If rotation is required to meet one of these criteria, it should be a 180 degree rotation about the X, Y, or Z axis, defined as follows:
| X | Rotate about Y | |
| Y | Rotate about Z | |
| Z | Rotate about X |
An axis of rotation or a principal axis of charge can be aligned with a Cartesian axis in one of two ways—either parallel or antiparallel, depending on the successive application of the following tests until a definite result is achieved:
The sum of the coordinates of the atoms of the highest atomic number on the axis must be positive.
The third moment of charge must be positive.
The sum of the projections of the atomic coordinates onto the reference axis must be positive.
The first atom with a non-zero projection on the reference axis must have a positive projection on that axis.
In the absence of any other rules, the principal axis corresponding to the largest principal moment of charge must be aligned with the highest priority Cartesian axis available. Individual point groups have specific considerations:
| Cs | The molecular plane must be made coincident with the XY plane. Note that although this convention conflicts with Mulliken’s suggestion, it is consistent with the character tables of Cotton and Herzberg. The molecule is then rotated about the Z axis according to the rules given below for Cn molecules. | |
| C2v | The molecular plane is placed in the YZ plane, following Mulliken’s recommendation for planar C2v molecules. The following tests are successively applied for non-planar molecules: (1) The mirror plane with the most atoms is put in the YZ plane; (2) The mirror plane with the most non-hydrogen atoms is put in the YZ plane; (3) The mirror plane with the lowest numbered atom is made coincident with YZ. Finally, the axes of charge rules are applied (as described above). | |
| Planar, D2h | Following Mulliken’s recommendation, the molecular plane is placed in the YZ plane. The molecule is rotated about the X axis so that the Z axis can pass through either the greater number of atoms, or, if this is not decisive, the greater number of bonds. | |
| Cn | Follow the rules for general symmetric top molecules. | |
| Ci | Translate but do not reorient. | |
| C1 | Translate but do not reorient. |
Symmetric top molecules are distinguished by having two of three moments of inertia equal. The third moment can thus be uniquely identified as the reference axis and the point group is analyzed by considering circular sets of atoms.
The following rules are applied for symmetric top molecules:
The unique axis is aligned with the Z axis.
A circular-set of atoms is composed of atoms lying in a plane which have the same atomic number, and are equidistant from a reference axis perpendicular to the plane. Atoms on the reference axis are not included in any circular-set. A circular-set of atoms is generated by a proper rotation axis.
The key atom in a symmetric top molecule is the atom with the lowest number in the key circular-set. The following tests are carried out successively to find the key circular-set:
Cn, Cnh, Sn: The molecule is rotated about the Z axis to maximize the number of pairs of heavy atoms parallel to the Y axis. If no such arrangement is satisfactory, then the key atom is placed in the YZ plane to give it a positive Y coordinate.
Dn, Dnh: One of the C2 axes is made coincident with the Y Cartesian axis. The tests described below are used to decide which C2 axis is so positioned.
Dnd, Cnv: One of the vertical planes is made coincident with the YZ Cartesian plane. The tests below are used to decide which plane is so positioned.
The following are tests for selecting among axes for the Dn, Dnh, Dnd, and Cnv molecules:
Maximize the projection of the key atom on the Y axis.
If two orientations give the maximum projection on the Y axis, select one with the maximum projection on the X axis.
For molecules contained in the XY plane, the standard axis orientation rules (see above) are applied to the X axis to complete the orientation specification.
Spherical top molecules are distinguished by having their equal moments of inertia and can be characterized by identifying spherical sets of atoms.
A spherical-set of atoms is composed of atoms which are equidistant from the origin and have the same atomic number. Spherical-sets should be ordered in terms of increasing distance from the origin and of increasing atomic number at any one distance. The key atom is the lowest numbered atom in the first spherical-set.
Although not generally the case, it is possible, with appropriate geometric constraints, to have D2d, D2h, or D2 molecules that are symmetric tops. Such molecules have three perpendicular two-fold axes that are aligned with the X, Y, and Z axes in accordance with the rules given above.
The following is a list of read-write files. Those that are permanently on the checkpoint file are marked with the letter P, and those that are temporarily on the checkpoint file are marked with the letter T. T files are saved for use in restarting an optimization or numerical frequency run, but are deleted when the job step completes successfully.
| Type | RWF | Description |
| P | 501 | Gen array. |
| P | 502 | /LABEL/—Title and atomic orbital labels. |
| 503 | Connectivity information (MxBond,0),NBond(NAtoms),IBond(MxBond,NAtoms),RBond(MxBond,NAtoms), where arrays are rounded to a multiple of IntPWP. | |
| 504 | Dipole derivative matrices (NTT,3,NAt3). | |
| P | 505 | Array of copies of /Gen/ from potential surface scan. |
| P | 506 | Saved basis set information before massage, uncontraction, etc. |
| P | 507 | ZMAT/ and /ZSUBST/. |
| P | 508 | /IBF/ Integral Bugger Format. |
| 509 | Incomplete integral buffer. | |
| T | 510 | /FPINFO/ Fletcher-Powell optimization program data. |
| P | 511 | /GRDNT/ energy, First and second derivatives over variables, NVAR. |
| P | 512 | Pseudo-potential information. |
| P | 513 | /DIBF/ integral derivative buffer format. |
| 514 | Overlap matrix, optionally followed by absolute overlap and absolute overlap over primitives. | |
| 515 | Core-Hamiltonian. There are four matrices here: H(α), the α core Hamiltonian; H(β), the β core Hamiltonian; G'(α), the α G' contribution to Fock matrix; G'(β), the β G' contribution to Fock matrix. H(α) and H(β) differ only if Fermi contact integrals have been added. The G' matrices are for perturbations which are really quadratic in the density (and hence have a factor of 1/2 in their contribution to the energy as compared to the true one-electron terms) but which are computed externally to the SCF. | |
| 516 | Kinetic energy and modifications to the α and β core Hamiltonian. These include ECP terms, Douglas-Kroll-Hess corrections, multipole perturbations and Fermi contact perturbations. The latter are used for calculations in which the nuclear and electronic Coulomb terms are computed together, such as the Harris functional and PBC calculations. For semi-empirical, holds the core Hamiltonian without nuclear attraction terms for use in the initial guess. | |
| 517 | Fermi contact integrals. | |
| 518 | Multipole integrals, in the order X,Y,Z,XX,YY,ZZ,XY,XZ,YZ,XXX,YYY,ZZZ,XYY,XXY,XXZ,XZZ,YZZ,YYZ,XYZ,XXXX,YYYY,ZZZZ, XXXY,XXXZ,YYYX,YYYZ,ZZZX,ZZZY,XXYY,XXZZ,YYZZ,XXYZ,YYXZ,ZZXY. | |
| T | 519 | Common /OptEn/—optimization control for link 109. |
| T | 520 | Electronic state: count and packed string (1+9 integers). |
| P | 521 | Electronic state: count and packed string (1+9 integers). |
| P | 522 | Eigenvalues, alpha and if necessary, beta. |
| 523 | Symmetry assignments. | |
| P | 524 | MO coefficients, real alpha. |
| P | 525 | (no longer used) |
| P | 526 | MO coefficients, real beta. |
| P | 527 | (no longer used) |
| T | 528 | SCF density matrix, real alpha. |
| T | 529 | (no longer used) |
| T | 530 | SCF density matrix, real beta. |
| T | 531 | (no longer used) |
| T | 532 | SCF density matrix, real total. |
| T | 533 | (no longer used) |
| T | 534 | SCF density matrix, real spin. |
| 535 | (no longer used) | |
| 536 | Fock matrix, real alpha. | |
| 537 | Fock matrix, imaginary alpha. | |
| 538 | Fock matrix, real beta. | |
| 539 | Fock matrix, imaginary beta. | |
| 540 | Molecular alpha-beta overlap (U), real. | |
| 541 | Molecular alpha-beta overlap (U), imaginary. | |
| T | 542 | Pseudo-potential information. |
| T | 543 | Pseudo-potential information. |
| T | 544 | Pseudo-potential information. |
| P | 545 | /ORB/ - window information. |
| 546 | Bucket entry points. | |
| 547 | Eigenvalues (double precision with window: always alpha and beta, even in RHF case). | |
| P | 548 | MO coefficients (double precision with window, alpha and if necessary beta). Complex if necessary. |
| 549 | Molecular orbital alpha-beta overlap, double precision with window. | |
| T | 550 | Potential surface scan common block. |
| T | 551 | Symmetry operaiton info (permutations, transformation matrices, etc.) |
| P | 552 | Character strings containing the stoichiometric formula and framework group designation. |
| T | 553 | Temporary storage of common/gen/ during FP optimizations. |
| T | 554 | Alternate starting MO coefficients, from L918 to L503, real alpha. Also MO coefficients in S-1/2 basis for L509 and rotation angles from L914 to L508. |
| 555 | Alternate starting MO coefficients, from L918 to L503, imaginary alpha. | |
| T | 556 | Alternate starting MO coefficients, from L918 to L503, real beta. Also MO coefficients in S-1/2 basis for L509 and rotation angles from L914 to L508. |
| 557 | Alternate starting MO coefficients, from L918 to L503, imaginary beta. | |
| 558 | Saved HF 2nd derivative information for G1, G2, etc. | |
| 559 | Common /MAP/. | |
| 560 | Core-Hamiltonian (a. o. basis) with 2 j - k part of deleted orbitals added in. (i.e. frozen core). | |
| P | 561 | External point charges or SCIPCM informations. |
| P | 562 | Symmetry operations and character table in full point group. |
| T | 563 | Integer symmetry assignments (α). |
| T | 564 | Integer symmetry assignments (β). |
| T | 565 | Lists of symmetry equivqlent shells and basis functions. |
| T | 566 | Unused in G09. |
| T | 567 | GVB pair information (currently dimensioned for 100 paired orbitals). |
| P | 568 | Saved hamiltonian information from L504 and L506. |
| P | 569 | Saved read-in window. |
| P | 570 | Saved amplitudes (IAS1,IAS2,IAD1,IAD2,IAD3; only IAS1 and IAD2 for closed-shell). |
| 571 | Energy weighted density matrix. | |
| 572 | Dipole-velocity integrals <Phi|Del|Phi'>, X, Y, and Z, followed by R × Del integrals (R × X, R × Y, R × Z). | |
| 573 | More SCIPCM information. | |
| T | 574 | /MSINFO/ Murtaugh-Sargent program data. |
| T | 575 | /OPTGRD/ Gradient optimization program data for L103, L115, and L509. |
| T | 576 | /TESTS/ Control constants in L105. |
| T | 577 | Symmetry adapted basis function data. |
| T | 578 | A logical vector indicating which MO’s are occupied. |
| T | 579 | NEQATM (NATOMS*NOP2) for symmetry. |
| T | 580 | NEQBAS (NBASIS*NOP2+NBas6D*NOp2) for symmetry. |
| T | 581 | NSABF (NBASIS*NOP2) for symmetry. Followed by matching integer character table, always (8,8). |
| T | 582 | MAPROT (3*NBASIS) for symmetry. |
| T | 583 | MAPPER (NATOMS) for symmetry. |
| P | 584 | FXYZ (3*NATOMS) cartesian forces. During PSCF gradient runs, there will be two arrays here: first the PSCF gradient, then the HF only component (needed for PSCF with HF 2nd deriv). |
| P | 585 | FFXYZ (NAT3TT) cartesian force constants (lower triangle). |
| T | 586 | Info for L106, L110, and L111. |
| T | 587 | L107 (LST) data. |
| 588 | Sx over cartesians in the ao basis. | |
| 589 | Hx over cartesians in the ao basis. | |
| 590 | F(x) over cartesians in the ao basis (all α, followed by all β for UHF) (without CPHF terms). | |
| 591 | U1(A,I) -- MO coefficient derivatives with respect to electric field and nuclear coordinates. | |
| 592 | Electric field and nuclear P1 (AO basis). | |
| 593 | Electric field and nuclear W1 (AO basis). | |
| 594 | Electric field and nuclear S1 (MO basis). | |
| 595 | Magnetic field U1(A,I) -- Del(X,Y,Z) then R × (X,Y,Z), 6 α followed by 6 β. | |
| 596 | Full MO Fock derivatives in the MO basis, including CPHF terms. | |
| P | 597 | Configuration changes for Guess=Alter. |
| 598 | User Name. | |
| 599 | Density basis set info: NDBFn, NVar, U0, DenBfn(4,NDBfn), ITypDB(NDBfn), Var(NVar), IJAnDB(NDBfn), IVar(4,NDBfn). | |
| 600 | Saved data for intra-link restart. | |
| P | 601 | Saved structures, and possibly forces and force constants along reaction path. All structures, then all forces, then all force constants. |
| 602 | Post-SCF two-particle density matrix. | |
| P | 603 | Density Matrices at various levels of theory. |
| T | 604 | common /drt1/ from drt program ... misc integer ci stuff, followed by variable dimension drt arrays. |
| P | 605 | Atomic charges from Mulliken Populations, ESP fits, etc. Bitmap followed by 0 or more NAtoms arrays. Bits 0/1/2/3/4 Mulliken/ESP-fit/Bader/NPA/APT. |
| 606 | SCF orbital symmetries in Abelian point group. Alpha and, if necessary, beta, full set followed by windowed set. | |
| 607 | Window’d orbital symmetries like rw 606 (always alpha and beta). | |
| 608 | IBF for sorted integrals (normally on SAO unit). | |
| 609 | Bit map for sorted integrals (normally on SAO unit). | |
| 610 | Sorted AO integrals (normally on SAO unit). | |
| 611 | NTT maps for sorted integrals (normally on SAO unit). | |
| 612 | Some 1E generators for direct CI matrix element generation. | |
| 613 | Some more 1E generators for direct CI matrix element generation. | |
| 614 | Configuration information for CAS-MP2. | |
| 615-616 | Used for CAS-MP2. | |
| 617 | Spin-orbit integrals. | |
| P | 618 | Nuclear coordinate third derivatives. |
| P | 619 | Electric field derivatives: 1 WP word bit map, dipole, dipole derivative, polarizability, dipole 2nd derivatives, polarizability derivatives, hyperpolarizability. |
| 620 | Magnetic field derivatives for GIAOs. | |
| 621 | Susceptiblity and chemical shift tensors. | |
| 622 | Partial overlap derivatives (<Mu|dNu/da>, NBasis*NBasis*NAt3). | |
| P | 623 | Born-Oppenheimer wavefunction derivatives (<Phi|d2Phi/dadb> for electronic Phi and a,b nuclear, NAt3TT). |
| 624 | Unused in G09. | |
| 625 | Expansion vectors and AY products from CPHF, in the order Y α, AY α, Y β, AY β. | |
| 626 | MCSCF MO 1PDM (NTT). | |
| 627 | MCSCF MO Lagrangian (NTT). | |
| 628 | MCSCF MO 2PDM (NTT,NTT) or NVTTTT. | |
| 629 | AO 2PDM (shell order). | |
| T | 630 | MCSCF information. |
| 631 | Post-SCF Lagrangian (TA, then TB if UHF). | |
| 632 | O*V*3*NAtoms, followed by O*V*NVar d2E/d(V,O)d(XYZ,Atom). | |
| P | 633 | Excited-state CI densities. |
| T | 634 | SCF Restart information (alpha, then possibly beta MOs). |
| P | 635 | CIS and CASSCF CI coefficients and restart information. |
| 636 | NBO analysis information. | |
| 637 | Natural orbitals generated by link 601. | |
| 640 | MCSCF data or CIS AO Tx’s for 2nd derivatives. | |
| 641 | MCSCF data for 2nd derivatives. | |
| 642 | MCSCF data for 2nd derivatives. | |
| 643 | MCSCF data for 2nd derivatives. | |
| 644 | MCSCF data for 2nd derivatives. | |
| 645 | MCSCF data for 2nd derivatives. | |
| 646 | MCSCF data for 2nd derivatives. | |
| 647 | MCSCF data for 2nd derivatives. | |
| 648 | MCSCF data for 2nd derivatives. | |
| 649 | Eigenvalue derivatives (non-canonical form even if done canonically). | |
| 650 | 2PDM derivatives, (LenTQ,NDeriv,ShellQuartet) order. | |
| 651 | Full U’s, canonical or non-canonical as requested. | |
| 652 | Generalized density derivatives for the current method (NTT,NDeriv,IOpCl+1). | |
| 653 | Lagrangian derivatives for the current method (NTT,NDeriv,IOpCl+1). | |
| 654 | Gx(Gamma). | |
| 655 | G(Gamma). | |
| 656 | Non-symmetric S1 and S2 parts of Lagrangian for MP2 or CIS second derivatives. | |
| 657 | t*Ix and t*Ix/D matrices from L811 for L1112. | |
| 658 | L(x) from L1111. | |
| 659 | MO correlated W for correlated frequencies. | |
| 660 | 2nd order CPHF results: Pia,xy, Sxy, Fxy (complete) all in MO basis, PSF α then PSF β if UHF. | |
| 661 | Computed electric field from L602. | |
| 662 | Points for electrostatic evaluation. | |
| T | 663 | Saved information for L117 and L124. |
| 664 | Spin projection data. | |
| P | 665 | Redundant coordinate information. |
| 666 | (no longer used) | |
| 667 | CIS AO Fock matrix. | |
| 668 | CIS Gx(T) matrices. | |
| 669 | Saved /ZMat/ and /ZSubst/ during redundant optimzations. | |
| P | 670 | New format basis set data (compressed /B/). |
| P | 671 | New optimization (L103/L104) data. |
| P | 672 | Unused in G09. |
| 673 | Global optimization data. | |
| 674 | ONIOM internal data. | |
| 675 | Saved files for LS during ONIOM. | |
| 676 | Saved files for MS during ONIOM. | |
| 677 | Saved files for LM during ONIOM. | |
| 678 | Saved files for HS during ONIOM. | |
| 679 | Saved files for MM during ONIOM. | |
| 680 | Saved files for LL during ONIOM. | |
| 681 | Saved files for HM during ONIOM. | |
| 682 | Saved files for ML during ONIOM. | |
| 683 | Saved files for HL during ONIOM. | |
| 684 | SABF information for DBFS: equivalent to files 577 and 581 for AOs. | |
| 685 | Cholesky U, or transformation to surviving basis functions. | |
| 686 | Cholesky U-1. | |
| 687 | Molecular mechanics parameters. | |
| 688 | Density in orthogonal basis (α spin) for ADMP or sparse SCF. | |
| 691 | Saved initial files during ONIOM (gridpoint 17, hence 674+17=691). | |
| 694 | Permutation applied to MOs for post-SCF symmetry. | |
| 695 | Magnetic properties. | |
| 696 | Saved magnetic field density derivatives. | |
| 698 | Saved initial structure during geometry optimization, in standard orientation, also used for constraints with the force constants following the structure. | |
| 699 | Density in orthogonal basis (β spin) for ADMP or sparse SCF. | |
| 700 | Saved /Mol/ for ONIOM. | |
| 701 | Saved Trajectory/IRC/Optimization history. | |
| 702 | Fit density for Coulomb. | |
| 703 | Fit density for Coulomb. | |
| 704 | Saved XC contribution to electric field F(xa) for polar derivatives. | |
| P | 710 | Basic PCM information. |
| P | 711 | Other PCM data. |
| P | 712 | Non equilibrium data for PCM. |
| 713 | Saved information for RFO with ONIOM microiterations. | |
| 714 | Saved model system information for ONIOM microiterations. | |
| 715 | Saved rigid fragment information for ONIOM microiterations. | |
| T | 716 | Saved copy of basis set data for counterpoise. |
| T | 717 | Saved copy of ECP data for counterpoise. |
| T | 718 | Saved copy of fitting basis for counterpoise. |
| 719 | Saved DiNa information. | |
| P | 720 | Saved DiNa information. |
| 721 | Frequency-dependent properties. | |
| 722 | Derivatives of frequency-dependent properties. | |
| 723 | Density fitting matrices (metrics). | |
| 724 | Density fitting basis (same format as /B/). | |
| 725 | DBF symmetry information (NEqDBF(NDBF,NOp2),NEqDB6(NDBF6D,NOp2)). | |
| 726 | DBF shell symmetry information (NEqDBS(NDBShl,NOpAll)). | |
| 727 | F(x)(P-Pfit) for density fitting second derivatives. | |
| 728 | PBC cell replication information. | |
| 729 | Alternate new guess during optimizations. | |
| 730 | Counterpoise input specification. | |
| 731 | Counterpoise intermediate data. | |
| 732 | Basis set for finite nuclei. | |
| 733 | PBC Cell scalars and integer cell indices. | |
| 734 | State-specific input parameters for SAC-CI. | |
| 735 | Excitation lables of SAC and SAC-CI. | |
| 736 | Eigenvalues and eigenvectors of SAC and SAC-CI. | |
| 737 | H matrices and their indices of non-zero elements used for SAC/SAC-CI. | |
| 738 | Saved atomic parameters for DFTB/EHTSC. | |
| 739 | Temporary storage for imaginary core Hamiltonian perturbations. | |
| 740 | Orbital information for SAC/SAC-CI gradients and PES by GSUM. | |
| 741 | MOD Orbital information for SAC gradients. | |
| 742 | Saved quadrature grid. | |
| 743 | Alpha Fock matrices in orthonormal basis for ADMP, also alpha HF Fock matrix for non-HF post-SCF. | |
| 744 | Beta Fock matrices in orthonormal basis for ADMP, also beta HF Fock matrix for non-HF post-SCF. | |
| 745 | K-integration mesh information. | |
| 746 | Eigenvalues and orbitals at all k-points. | |
| 747 | Information for external low-level calculations for ONIOM. | |
| 748 | TS vector information for ONIOM TS optimizations. | |
| 749 | Conical intersection information for ONIOM. | |
| 750 | Not used in G09. | |
| 751 | Temporary storage for SO ECP integrals. | |
| 752 | Pseudo-canonical MO Fock matrix for ROMP and ROCC. | |
| 753 | Data for FD polar derivatives. | |
| 754 | Saved PCM charge derivatives. | |
| 755 | PCM inverse matrices. | |
| 756 | Charge information for ONIOM. | |
| 757 | MO:MO embedding charge data for L924. | |
| 758 | Derivatives of embedding charges, when computed explicitly. | |
| 759 | Basis set info for density embedding. | |
| P | 760 | Full set of pseudocanonical orbitals for RO. |
| 761 | Charges from external PCM iterations (both L117 and L124). | |
| 762 | Saved weights for non-symmetric Mulliken analysis. | |
| 763 | File for FC/HT integrals. | |
| 764 | File for FC/HT integrals. | |
| P | 765 | Saved normal modes. |
| 766 | Saved QuadMac vectors (temporary). | |
| 767 | CIS coefficients reordered by symmetry. | |
| 768 | Semi-empirical parameters. | |
| 769 | Saved MOs during numerical differentiation. | |
| P | 770 | Saved ground-to-excited state energies and transition moments. |
| 771 | EOM iteration information. | |
| 772 | Symmetry operations and character table in Abelian point group. | |
| 989 | Multi-step job information (1000 reals and 2000 integers). | |
| 990 | KJob info in some implementations. | |
| 991 | Holds file names, ID’s and save flags. | |
| 992 | Used for link substitution information in some implementations. | |
| 993 | COMMON /INFO/ | |
| 994 | COMMON /PHYCON/ | |
| 995 | COMMON /MUNIT/ | |
| 996 | COMMON /IOP/ | |
| P | 997 | COMMON /MOL/ |
| P | 998 | COMMON /ILSW/ |
| 999 | Overlay data. |
Last update: 21 April 2014