Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2026-01-15 19:04:38
- Ainosya
- Member
- Registered: 2024-12-06
- Posts: 18
Using Cube Files to Obtain r2ψ2 of specific MO
Hello,
I would like to ask for clarification regarding how to obtain r2∣ψ∣2 from a cube file of a specific molecular orbital using Multiwfn.
Usng a .fchk file that I generated using G16, I followed this sequence in Multiwfn:
200->5->1->44->ha->0->1
And obtained the following results: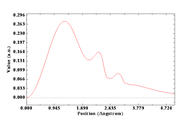
However, when using a cube file format generated by G16 with the command:
cubegen 1 MO=all Octamer.fchk Octamer_MO.cube 80 h
and loading MO 41 into Multiwfn, I followed this sequence:
200 -> 5 -> 1 -> 1 -> 0 -> 1
and obtained: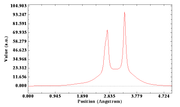
Since I am specifically loading MO 41 (which corresponds to the HOMO), I would like to know how I can generate the same first plot using only a cube file. Although this may seem counterintuitive, I want to use the cube file format because my own software generates only cube files, and I am currently using Gaussian 16 as a sanity check.
Thank you for your help
Last edited by Ainosya (2026-01-15 20:32:36)
Offline
#2 2026-01-20 06:06:16

Re: Using Cube Files to Obtain r2ψ2 of specific MO
Hello,
First there is a big error: If you select "1 Electron density (rho)" in "1 Select real space function", then electron density will be calculated by Multiwfn based on wavefunction, however, cube file cannot provide wavefunction to Multiwfn (it can only provide grid data to Multiwfn. Please carefully check Section 2.5 of Multiwfn manual for details).
Please solve this issue first.
By the way, when you hope to use an external grid data to define the function to be studied/plotted, you should set "iuserfunc" in settings.ini to -1 (linear interpolation) or -3 (B-spline interpolation), then load cube file to Multiwfn after launching it, and then select "100 User-defined function" as the function to be studied/plotted in the real space function selection interface.
Best,
Tian
Offline