Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2025-10-06 04:22:00
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Error: The promolecular density for the element with index of 113 has
Dear aal
When I was trying to get aNCI for my simulation (filename.xyz) I got an error message 'Error: The promolecular density for the element with index of 113 has not been supported yet!'
I followed the steps as mentioned in the article "Using Multiwfn to perform aNCI analysis to graphically study protein-ligand interactions in dynamic process" http://sobereva.com/591
In details:
20
3
1,1001
11
72-130 (atomic number of the LIG 'the number of the first and last atoms of the LIG as in the .pdb file' Note, I got the .pdb file just after I saved the .xyz file)
4.5
0.17
Coordinate of origin in X,Y,Z is 47.309292 -47.095754 -21.081785 Bohr
Coordinate of end point in X,Y,Z is 79.269292 -15.645754 12.068215 Bohr
Grid spacing in X,Y,Z is 0.170000 0.170000 0.170000 Bohr
Number of points in X,Y,Z is 189 186 196 Total: 6890184
Calculating averaged density, gradient and Hessian of density
Error: The promolecular density for the element with index of 113 has not been supported yet!
Error: The promolecular density for the element with index of 113 has not been supported yet!
Error: The promolecular density for the element with index of 113 has not been supported yet!
Error: The promolecular density for the element with index of 113 has not been supported yet!
As you see the Error comes out.
Kindly, try to help me to solve this.
Regards
Offline
#2 2025-10-06 07:05:56

Re: Error: The promolecular density for the element with index of 113 has
Please make sure that element names rather than atom names appear in your .xyz file. If atom names were used, then Multiwfn sometimes cannot correctly guess elements. For example, CA may refer to Calcium atom or alpha-C atom in AMBER forcefield.
Alternatively, as mentioned in my blog article, provide a .pdb file with same name as .xyz file in current folder, and make .pdb file record correct element names, then Multiwfn will automatically read element names from the .pdb file when loading .xyz file.
Offline
#3 2025-10-06 09:05:42
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
I have already added a .pdb file with the same name as .xzy
So, as you mentioned, I have to record correct element names in that .pdb file. so to change CA into carbon 'only'
Note, the .pdb file it too large
Am i right?
Offline
#4 2025-10-06 09:08:15
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
I mean in my last reply that the .pdb file contains atoms of the Ligand and surrounding amino acids within 3.5 A and some H2O molecules.
the total number is 320 atoms. however, in the .pdb file these atoms repeated more and more for several repeated times.
Offline
#5 2025-10-08 00:28:12
Re: Error: The promolecular density for the element with index of 113 has
You need to make sure that the .pdb file records element names rather than atom names, otherwise Multiwfn still cannot aceept correct element information. In my blog article I have already mentioned how to realize this: (simply translated by Google)
One important point to note is that aNCI analysis requires knowing the element corresponding to each atom; otherwise, it's impossible to construct the promolecular density. Therefore, the xyz trajectory file used for aNCI analysis should contain the element name of each atom (as standard xyz files should). Otherwise, Multiwfn will guess based on the atom name, which can easily lead to errors. For example, it might be impossible to distinguish whether CA is calcium or alpha carbon. Since md_fix_nowat.gro records the atom names used during the GROMACS dynamics run, and this format doesn't record element information, the atom names are also used in the cluster.xyz file saved above. So how can Multiwfn correctly obtain element information? One approach is to manually replace the atom names in cluster.xyz with element names. This is easy for simple cases like the water box, where there are only three types of atom names, so replacing them three times is sufficient. However, this is clearly too laborious for the current system. Therefore, Multiwfn has designed a special rule: if there is a file in the current directory with the same name as the loaded xyz file but with the suffix pdb, the element name will be preferentially read from the element column in the pdb file. However, if the pdb file doesn't provide element names for certain atoms, Multiwfn will still guess them based on the atom names in the xyz file, automatically removing any numbers from the names. Therefore, the goal now is to generate a pdb file containing element information for the corresponding local region. The steps are as follows.
Run gmx editconf -f md_fix.tpr -o md_fix.pdb to convert the current dynamics tpr file into a pdb file. Because the tpr file contains element information for the protein, the resulting pdb file also contains element information for the protein portion, which is the last column of the newly generated md_fix.pdb. However, when you open md_fix.pdb and locate the position of the MOL residue, you will see that the ligand portion of the resulting pdb file lacks element information. However, this isn't a problem in this case, because the atom names for the ligands in cluster.xyz are all names like C1, N8, and H13. The characters after the numbers directly match the element names. Therefore, there's no need to manually add the element names for the ligands in md_fix.pdb, nor is there a need to change all the ligand atom names in cluster.xyz to element names. (However, for other systems, it's best to change one or the other to be safe.)
Note: Incidentally, let's discuss the circumstances under which element information is recorded in the PDB. In [atomtypes], if the atom type name is followed by the element's periodic table number, then atoms using this atom type in the TPR will have element information, and thus, will also have element information after conversion to the PDB. The GROMACS built-in protein force field defines element numbers in [atomtypes] in ffnonbonded.itp, so the protein portion of the converted PDB will always have elements. For small molecules, it depends on the type of force field you use.
Offline
#6 2025-10-09 15:31:19
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Dr. Tian Lu
I am greatly appreciating your reply, I would like to inform you that I have succeeded to get the run work. I will attach you the result picture since I got so many red color around my ligand, keeping in mind that I used VMD to choose residues around my ligand within 3.5 A and remove all water molecules and others protein residues and also ions, and grid spacing was 0.13 bohr.
how can I solve this?
By the way, do you think that analyzing IGM (Independent Gradient Model) may get better information than aNCI?
Thanks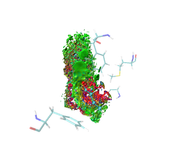

Offline
#7 2025-10-10 06:13:01
Re: Error: The promolecular density for the element with index of 113 has
IGM and aNCI are completely different, the former is used to analyze single frame, while the latter is used to characterize averaged interactions during MD.
aNCI is fully out-of-date. I STRONGLY suggest using the amIGM method proposed by me this year, see:
Tian Lu, Graphically Revealing Weak Interactions in Dynamic Environments Using amIGM Method, Struct. Bond. (2025) https://doi.org/10.1007/430_2025_95
amIGM example has also been provided in the latest version of Multiwfn manual (Section 4.20.13).
amIGM is the best method for characterizing dynamically averaged weak interactions.
In addition, even if you are only interested in studying single-frame, mIGM is also a much better choice than IGM. mIGM was also proposed in the paper mentioned above and can be realized via Multiwfn (see Section 4.20.12 of manual for example).
More information and comparison can be found in my recent review:
Tian Lu, Visualization Analysis of Covalent and Noncovalent Interactions in Real Space, Angew. Chem. Int. Ed., 137, e202504895 (2025) DOI: 10.1002/anie.202504895
Offline
#8 2025-10-10 07:22:08
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Tian
Can you share me the above-mentioned manuscripts
Tian Lu, Graphically Revealing Weak Interactions in Dynamic Environments Using amIGM Method, Struct. Bond. (2025) https://doi.org/10.1007/430_2025_95
Tian Lu, Visualization Analysis of Covalent and Noncovalent Interactions in Real Space, Angew. Chem. Int. Ed., 137, e202504895 (2025) DOI: 10.1002/anie.202504895
As i don't have access to get it both.
Offline
#9 2025-10-10 08:27:28
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Lian
One important thing, after I got the selected coordinates for a ligand and the surrounding amino acids in .xyz file for about 1000 frames usinf the following (same residue as within 3.5 of resname LIG or (not protein and not waters and not ions and not ion)) in VMD, when I used to open .xyz file with notepad++, I got that there are some water molecules and one Ion, so I remove all water molecules and ions from the .xyz file.
So the .xyz file will have only atomic coordinates for amino acids and ligand
Is this, okay? or I should not remove the water molecules?
Kindly, reply for this matter.
Thanks
Offline
#10 2025-10-10 08:58:44
Re: Error: The promolecular density for the element with index of 113 has
Dear Tian
Can you share me the above-mentioned manuscripts
Tian Lu, Graphically Revealing Weak Interactions in Dynamic Environments Using amIGM Method, Struct. Bond. (2025) https://doi.org/10.1007/430_2025_95
Tian Lu, Visualization Analysis of Covalent and Noncovalent Interactions in Real Space, Angew. Chem. Int. Ed., 137, e202504895 (2025) DOI: 10.1002/anie.202504895
As i don't have access to get it both.
Please show me your E-mail.
Offline
#11 2025-10-10 09:00:27
Re: Error: The promolecular density for the element with index of 113 has
Dear Lian
One important thing, after I got the selected coordinates for a ligand and the surrounding amino acids in .xyz file for about 1000 frames usinf the following (same residue as within 3.5 of resname LIG or (not protein and not waters and not ions and not ion)) in VMD, when I used to open .xyz file with notepad++, I got that there are some water molecules and one Ion, so I remove all water molecules and ions from the .xyz file.
So the .xyz file will have only atomic coordinates for amino acids and ligandIs this, okay? or I should not remove the water molecules?
Kindly, reply for this matter.Thanks
If you only want to study interaction between amino acids and ligand, you can remove all other parts.
Offline
#12 2025-10-11 03:33:13
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Lian
As you mentioned that NCI is out-of-date and the best is
amIGM is the best method for characterizing dynamically averaged weak interactions. okay
if i will study for a single compound, should I use NCI or mIGM
Thanks, here you are my email address
melhenawy111@gmail.com
Offline
#13 2025-10-11 07:56:04
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Lian
Here you can find the result of mine, as I understand from reading-out the manual regarding mIGM.
1- From the cluster analysis result I got the selected amino acid residues near the ligand and then save the file in .xyz containing one frame only and upload it to Multiwfn, then follow the steps of mIGM.
The results contain: 1) blue color for strong interactions like H-bond. 2) Green color for the noncovalent interactions like Van der Waals. 3) Red color for allosteric.
Is this result correct and my conclusion right?
Thanks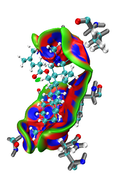

Offline
#14 2025-10-11 13:44:14
Re: Error: The promolecular density for the element with index of 113 has
The maps look very weird. Perhaps the isovalue was not properly set (should be too small). Without your structure file I cannot reproduce your result and clearly answer this question.
Offline
#15 2025-10-12 09:19:06
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Dr. Lu
Thanks for your reply, How can i send you the file?, send me your email address.
Kindly. send me copy of the recent two articles for mIGM and amIGM. my email address is; melhenawy111@gmail.com
Regards
Offline
#17 2025-10-12 13:42:15
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Lu
I did not receive your email message, kindly try to resend the articles again.
emails:
melhenawy111@gmail.com
mm.mohammed@nrc.sci.eg
by the way check the following two images and send me your comments.
Thanks 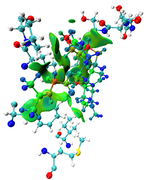
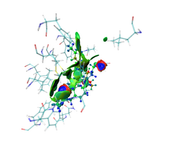
Offline
#19 2025-10-12 17:25:10
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Thanks, Dear Lu
I will check email.
Thanks
Offline
#20 2025-10-13 12:52:08
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Dear Lu
What is the reason for;
1- I finished mIGM analysis and trying to get the (Evaluate contribution of atomic pairs and atoms to inter-fragment interaction (atomic and atomic pair delta-g indices as well as IBSIW index))
2- Unfortunately, I couldn't get the same colors deep green for good interactions and blue is for atoms away from interaction.
I did the same as you mentioned;
First, plot color-filled isosurfaces using the IGM_inter.vmd script as mentioned earlier. After that, we need to remove the default representation showing molecular structure, so we enter "Graphics" - "Representation", choose the first term (its current style is CPK), click "Delete Rep" button. Then we drag the previously generated atmdg.pdb into VMD main window to load it. In this file the “occupancy” field records intermolecular interactions, to graphically exhibit its value for every atom, we should let VMD color the atoms according to their occupancy property. We enter "Graphics" - "Representation" again, set "Drawing method" to CPK, and set "Coloring method" to “Occupancy”,
then click "Trajectory" tab, set upper limit of color scale to 50 and press ENTER button, now the system in graphical window should look like below.
Note
Atomic pair delta-g indices and percentage contributions (zero terms are not shown)
5 351 : 19.556075 ( 2.50 % )
3 351 : 17.894879 ( 2.29 % )
193 350 : 16.489099 ( 2.11 % )
178 350 : 15.737952 ( 2.01 % )
12 350 : 14.503133 ( 1.85 % )
check the attached file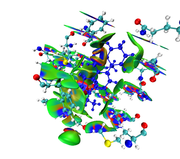
Offline
#21 2025-10-13 12:55:35
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
as you see in the last image there are some isosurfaces appeared within the amino acid residues, even though, I put 'Input index of the atoms to define a fragment, e.g. 2,3,7-10' around that for Ligand only.
Kindly, check and comment
Offline
#22 2025-10-19 08:02:45
- melhenawy111
- Member
- Registered: 2025-10-06
- Posts: 15
Re: Error: The promolecular density for the element with index of 113 has
Kindly, check your email
Offline
#23 2025-10-19 18:41:50
Re: Error: The promolecular density for the element with index of 113 has
Hello,
Your map is not very satisfactory. I suggest you inputting the following commands in Multiwfn after loading the .xyz file
20
-10
2
279-349
c
11
279-349
4 A
0.15
3
Then use igm_inter.vmd to render the cube files, and set isovalue to 0.003 and properly modify representation, then image looks nice:
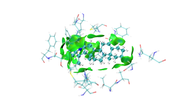
Offline