Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2025-09-17 04:30:31
- Hengyuan
- Member
- Registered: 2025-09-17
- Posts: 9
Plot electron density along a line with fixed atom positions
Dear Tian,
I am trying to plot the electron density of a water dimer along the O-O bond. The geometry of this is as follows in Q-Chem input format with unit in Angstrom:
$molecule
0 1
--
0 1
O 0.000000 0.000000 0.000000
H 0.480763 -0.756792 -0.334637
H 0.480763 0.756792 -0.334637
--
0 1
O 0.000000 0.000000 2.900000
H -0.899690 0.000000 3.225430
H -0.089397 0.000000 1.947180
$end
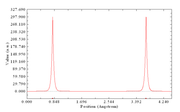
I was able to generate the line density along the O-O bond with Multiwfn, however, it seems like the molecule was shifted so the nuclei aren't in the initial position as input. I was able to confirm this conjecture by comparing the z coordinate and the X position in the curve graph in the saved line.txt file. Is there a way to fix the position of the molecule when generating the line density plot? Thank you in advance for your time!
Offline
#2 2025-09-17 10:26:56

Re: Plot electron density along a line with fixed atom positions
Hello,
If you choose two atomic nuclei to define the line to be plot in Multiwfn, then the value of X-axis will corresponds to the distance from starting point, which is irrelevant to Cartesian coordinate of the atoms.
Offline
#3 2025-09-17 18:10:21
- Hengyuan
- Member
- Registered: 2025-09-17
- Posts: 9
Re: Plot electron density along a line with fixed atom positions
Dear Tian,
Thank you for your reply. I should have chosen "Input coordinate of two points to define a line", or set the extension distance to 0 before using "Input index of two atoms to define a line". Thank you again for your help.
Offline