Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2024-04-18 01:27:05
- prvnpa4
- Member
- Registered: 2023-12-26
- Posts: 3
BLA Calculations are not completing
Dear Prof. Tian Lu,
I am trying to calculate the bond length alteration values for a thiophene system.
I used the optimized geometry with Multiwfn (200.18) for that. When I was asked to choose the rings and end atoms, I used appropriate details. For example, in the case below, I chose 1-39 and 3 and 38 as end atoms.
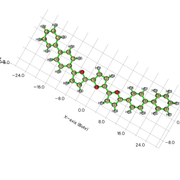
However, once I input the values, the calculations are not complete (I waited up to 5 hours). I tried using both Gaussian and ORCA log/output files as well as XYZ files. I have also tried using fchk as well as molden files converted from G09 and ORCA.
I am not sure whether I am doing things properly. Would you please kindly assist me in this regard?
Thanks and regards,
Praveen
Offline
#2 2024-04-18 03:03:48

Re: BLA Calculations are not completing
Dear Praveen,
You inputted atomic indices in a wrong way. Multiwfn needs to determine the path (atomic chain) to be studied, however based on your input Multiwfn cannot infer a unique path. It is best to manually input all indices of atoms according to connectivity from beginning to ending atoms, for example, 3,2,1,6,9,8 ... 31,32,35,34,39,38
Best,
Tian
Offline
#3 2024-04-18 06:33:02
- prvnpa4
- Member
- Registered: 2023-12-26
- Posts: 3
Re: BLA Calculations are not completing
Dear Praveen,
You inputted atomic indices in a wrong way. Multiwfn needs to determine the path (atomic chain) to be studied, however based on your input Multiwfn cannot infer a unique path. It is best to manually input all indices of atoms according to connectivity from beginning to ending atoms, for example, 3,2,1,6,9,8 ... 31,32,35,34,39,38
Best,
Tian
Dear Prof. Lu,
Thank you very much. Now, I am able to calculate the BLA values.
Kind regards,
Praveen
Offline