Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2023-12-14 11:09:13
- saeed_E
- Member
- Registered: 2019-12-21
- Posts: 348
HOMO and LUMO for open-shell systems
Dear Tian,
Please consider an open-shell system with, for instance, charge of zero and multiplicity of 5. The highest singly occupied ALPHA orbital should be taken as HOMO while the next empty orbital over the highest singly occupied ALPHA orbital should be taken as LUMO. Are you quite agree with me?
Sincerely yours,
Saeed
Offline
#2 2023-12-14 12:10:06

Re: HOMO and LUMO for open-shell systems
Dear Saeed,
No. In this case, beta-LUMO should have lower energy than alpha-LUMO. Certainly alpha-LUMO cannot be regarded as the LUMO of the system in common sense.
Tian
Offline
#3 2023-12-14 13:17:27
- saeed_E
- Member
- Registered: 2019-12-21
- Posts: 348
Re: HOMO and LUMO for open-shell systems
Dear Tian,
Too many thanks for your kind attention to prompt reply with highly valuable comments. Please, to avoid any confusions, let me explain you my understanding through an attached picture. In this picture related to the MOs of CrCl2 (charge=0, multiplicity=5), the HOMO and LUMO levels are characterized considering your nice comments. 1) are these levels correct?
2) Indeed, for any purpose for which the HOMO and LUMO levels of this compound are requested such as computing DFT reactivity indices (chemical hardness, electronic chemical potential, etc.) or the HOMO-LUMO gap between this compound bound to a dienophile such as acroleine and a given diene in a Diels-Alder reaction the distinguished orbitals on the picture should be used to specify normal/inverse electron demand type. Correct?
Sincerely,
Saeed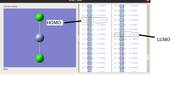
Last edited by saeed_E (2023-12-14 13:18:51)
Offline
#4 2023-12-14 13:55:14
Re: HOMO and LUMO for open-shell systems
1 correct
2 Never use MO to compute the reactivity indices. Always calculate them using their standard definitions, namely evaluate based on electronic energy of different charged states.
Offline
#5 2023-12-14 14:10:36
- saeed_E
- Member
- Registered: 2019-12-21
- Posts: 348
Re: HOMO and LUMO for open-shell systems
Dear Tian,
Thank you very much.
Sincerely,
Saeed
Offline