Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2023-04-29 13:40:37
- glauciobf
- Member
- Registered: 2023-04-29
- Posts: 2
Help for using ELF in Coordination Compounds.
I am evaluating the use of Multiwfn for comparative work on QTAIM and ELF, as found in recent papers on coordination compounds. So, I did a test and based on the models from the Multiwfn manuals. However, I have a doubt, when identifying the attractors in the ELF analysis that are generated from the electronic density. I do not identify points on the bond axis, as in the case of the QTAIM analysis. In fact, what I identify and what I have the impression is that they are the isolated pairs of the O atom of the water molecule and the orbitals of the metallic ion (Zn2+). Is this right? Because when trying to analyze this way, the result indicates a very low interaction at these points. Could anyone give any tips?
Do I need to make any corrections for proper analysis of attractors in the M-O bond?
The aquacomplex [Zn(OH2)6]2+ was optimized with Functional M06L2X and base function Def2tzvp.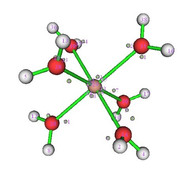
Offline
#2 2023-04-30 03:41:15

Re: Help for using ELF in Coordination Compounds.
I don't understand your meaning "ELF analysis that are generated from the electronic density". Commonly, ELF is calculated based on wavefunction instead of electron density. Although Multiwfn also supports a special variant of ELF, which can be calculated fully based on electron density, its quality is significantly poorer than the original ELF definition and thus rarely used if wavefunction is available.
Here I assume that you are studying the system using standard ELF definition. The ELF attractors shown in your map is not unexpected, I suggest you plotting an ELF isosurface map via main function 5 of Multiwfn, and compare it with the attractor map, and meantime gradually adjust isovalue of the ELF isosurface map, you will better recognize the distribution character of ELF.
Offline
#3 2023-04-30 16:50:01
- glauciobf
- Member
- Registered: 2023-04-29
- Posts: 2
Re: Help for using ELF in Coordination Compounds.
Dear Tian Lu.
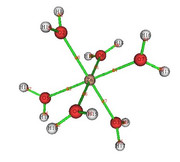
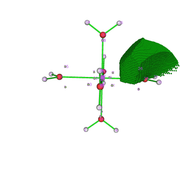
I fully understood the effect of the grid value on the analysis. Thanks. I reevaluated the data and it made more sense. I still have two doubts: 1) V(Zn,O) in the figure that describes the attractor 19 (fig1) and the attractors 26 and 9 would the V(O) represent the isolated pairs (fig 2 is atractor 26 which is close to atractor 9)? The electron density analysis matches this assessment.
2) Is there a DI value for the BPC obtained by QTAIM? Should this be calculated using the BPC coordinate by redoing the basin analysis in the option Study source function in AIM basins?
Thank you for your attention.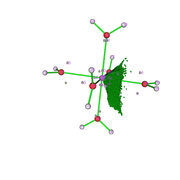
Offline
#4 2023-04-30 19:01:54
Re: Help for using ELF in Coordination Compounds.
1 In my view, both the basins may be assigned as V(Zn,O), because both of them contribute to the Zn-O coordination bond.
2 Do you mean "BCP"? (bond critical point) If yes, please note that DI is defined for two subregions of your system, for example, you can say DI between two basins. DI value cannot be calculated just for a BCP. However, if what you really want is to calculate contribution of electrons in a ELF basin to BCP via integrating source function in the basin, it is fully feasible via Multiwfn.
Offline