Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2022-08-22 14:58:13
- Aerael
- Member

- From: Germany, Bavaria, Munich
- Registered: 2020-10-08
- Posts: 16
Bug in non-singlet NAdO orbcomp analysis
[EDIT: ISSUE SOLVED, IT WAS A USER ERROR, NOT A BUG. FOR A WORKAROUND, READ THE LAST FEW REPLIES!]
Dear Tian Lu,
the orbcomp routine (after 17 basin analysis is done --> 11) can be used to evaluate the natural adaptive orbitals of a .fchk file within the QTAIM basins of the whole electron density contained within the same file. This works flawlessly for simple cases and I use this method.
Unfortunately, for non-singlet transition metal cases, this results in faulty behaviour.
The percentages per spin orbital do not add up to 100.
E.g. for CH4+, all looks perfectly in line with the shape of the NAdOs, and also for other non-singlet systems.
For transition metal systems, it fails. If you calculate a triplet and a singlet state of the same system, the singlet NAdOs orbcomp analysis will result in numbers that match the shapes and add up, while the triplet calculation will not.
Please have a look yourself at the NAdO orbcomp of any non-singlet transition metal system.
I can also email you two examples if you would like.
Thanks so much as always and best wishes and health to you and your family!
Daniel
Last edited by Aerael (2022-09-20 11:22:46)
Daniel, PhD student at LMU Munich, Germany under Prof. Dr. Klüfers. Special interest: transition metal nitrosyl complexes.
Offline

#3 2022-08-25 04:45:01
Re: Bug in non-singlet NAdO orbcomp analysis
I have received your files and replied you via E-mail. This is not bug, but due to inadequate quality of your wavefunction. If you hope to perform NAdO analysis based on AIM partition, the wavefunction quality should not be too poor, which may lead to artificial non-nuclear attractors and hinders NAdO analysis. 6-31G* is often too small for some transition-metal coordinates.
Offline
#4 2022-08-25 13:16:15
- Aerael
- Member
- From: Germany, Bavaria, Munich
- Registered: 2020-10-08
- Posts: 16
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Tian Lu,
I have not received an email reply from you (also not in spam folder). Still, this is incorrect.
My used level of theory is and was NOT B3LYP / 6-31g(d) like it is wrongly stated in the respective .fchk files.
We use def2-tzvp + bp86/d3 (+ rijcosx + def2/j which is virtually impactless) and CPCM in ORCA 5.0.3. This combination is well suited to accurately describe partial charges, effective oxidation states, geometries, and vibrational modes of TMCs, and for our systems (nitrosyl transition metal complexes) it is more accurate than using Minnesota or Head-Gordon functionals and a variety of other meta hybrids, hybrids and pure functionals.
This is cool, so we can resolve two bugs at once, haha. ![]()
The .fchks were exported from an orca 5.0.3 wave function read into MultiWFN directly using the orca_2mklpath variable in the settings.ini.
I do not know why the level of theory is read in wrongly by MultiWFN, but it happens for all Orca wave functions, no matter if NAdO or usual, and it also happened independent of Orca main version (tried 4.2.1 and 5.0.3). But this is not important to me, honestly.
Anyway, I can reproduce the NAdO orbcomp error for an UKS singlet calculation, for which the respective RKS calculation of identical quality does NOT show the faulty behaviour. (I used the singlet example I sent you.) UKS and RKS give close to numerically identical results for charges and effective oxidation states based on the same AIM analysis from which I produce the NAdOs, so the AIM analysis is not at fault here.
I was also able to reproduce the error for a def2-qzvpp bp86/d3 (rijcosx def2/j CPCM) calculation of the triplet example I sent you.
I used tight optimisation and SCF convergence criteria, and the WF is not the same as in the optimisation runs, but from a subsequent single point.
Hence, the wave function quality is not to blame either. This is clearly a bug in how unrestricted .fchks are handled by the orbcomp subroutine (EDIT: or how the unrestricted nado.mwfn files are exported, or how unrestricted NAdO-.fchks are exported after being read in from the unrestricted nado.mwfn file).
Thanks for getting at it again! ![]()
We all appreciate all your hard work here!
Last edited by Aerael (2022-08-25 13:50:39)
Daniel, PhD student at LMU Munich, Germany under Prof. Dr. Klüfers. Special interest: transition metal nitrosyl complexes.
Offline
#5 2022-08-25 15:09:40
Re: Bug in non-singlet NAdO orbcomp analysis
My E-mail was rejected by your mail server, now I attach my E-mail content here:
Dear Daniel,
I found it is not a bug, but possibly due to inadequate quality of your wavefunction.
The screenshot is distribution of attractors located for triplet_nados_BUG.fchk, there are many non-nuclear attractors (NNA), some of them are highlighted by pink circles. In this case, there is no one-to-one correspondence between basins and atoms, and thus NAdO doesn't properly work.
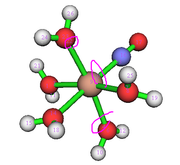
I recalculated your system using much better basis set def2-TZVP, the input file and the resulting .fchk file are provided. If you perform basin analysis for this wavefunction, you will not see any unexpected NNA, and then NAdO should work.
Gaussian input file of my calculation:
%chk=t.chk
# ub3lyp/def2tzvp
Generated by Multiwfn
2 3
Sc -0.85507463 1.63593853 2.72702256
N -0.81421198 3.54375878 3.39373225
O -0.78150185 4.67299901 3.78685139
O -0.87279129 -0.42351288 1.97267754
H -1.67216251 -0.92663181 1.72999795
H -0.12136462 -1.04459666 1.95907772
O -2.98379670 1.53738536 2.22450725
H -3.67007750 1.52126336 2.91905173
H -3.34810852 2.06316932 1.48659056
O 1.25619784 1.18981787 3.06555344
H 1.96735404 1.80144469 2.79513419
H 1.56490917 0.74094707 3.87592670
O -0.44754365 2.20440673 0.69269487
H -0.40733112 1.58868856 -0.06348351
H -0.37798753 3.11142335 0.34102867
O -1.34052091 0.79547839 4.65052002
H -1.56075783 -0.13936373 4.82500885
H -1.54380063 1.30004898 5.46034148Also note that when there are NNAs, the data in orbcomp.txt do not sum up to 100% because NNAs are ignored when printing result.
Best,
Tian
Offline
#6 2022-08-25 16:00:51
- Aerael
- Member
- From: Germany, Bavaria, Munich
- Registered: 2020-10-08
- Posts: 16
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Tian,
thank you so much.
I see, so for some reason, between importing the correct orca WF (def2-tzvp / bp86) contained in the .gbw and the export of my NAdO wave function either via nado.mwfn (or the subsequent conversion to .fchk), the basis set gets reduced. That is bad news for me.
I will test some more, and when I find out where in the chain of conversion the error occurs, I will open a new thread.
Until then!
Daniel, PhD student at LMU Munich, Germany under Prof. Dr. Klüfers. Special interest: transition metal nitrosyl complexes.
Offline
#7 2022-08-26 09:36:53
- Aerael
- Member
- From: Germany, Bavaria, Munich
- Registered: 2020-10-08
- Posts: 16
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Tian Lu,
I now know where things go wrong. After exporting the NAdO.mwfn from the correctly built AOM, I leave MultiWFN.
I then read in the exported NAdO.mwfn (to have access to NAdOs), and do a basin analysis from that file. This is the step where all goes wrong.
The resulting basin analysis is indeed inaccurate because it is done from the NAdO density, but only gives out an explicit warning if one tries to expert the AOM.
I instead tried loading in the correct basins (from a basin.cub of the correct AOM) after I open the NAdO.mwfn in MultiWFN, but failed.
I also tried to built the correct basins, and then load in in the NAdOs from their NAdO.mwfn, but also failed.
Could you please explain to me how I can get an orbcomp analysis of the NAdOs, but within normal SCF density AIM basins?
(I'm sending you the orca5.0.3 .gbw from the triplet def2-tzvp / bp86/d3 calculation via email.)
All the best,
Daniel
Daniel, PhD student at LMU Munich, Germany under Prof. Dr. Klüfers. Special interest: transition metal nitrosyl complexes.
Offline
#8 2022-08-26 14:46:11
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Daniel,
There is a workaround:
First, load original wavefunction (i.e. the wavefunction file directly produced by quantum chemistry code) into Multiwfn, use main function 5 to calculate grid data of electron density and export to density.cub file.
Reboot Multiwfn, load NAdO.mwfn, then enter main function 13, now you can input path of density.cub to load it into memory. Then return to main menu and enter basin analysis module. When you choose "1 Generate basins and locate attractors", you will find this time you can choose option "2: Generate the basins by using the grid data stored in memory", after selecting it the basins will be generated based on the grid data of correct total electron density (which comes from density.cub). Then you can normally use option "11 Calculate orbital compositions contributed by various basins" to evaluate orbital composition for the NAdO orbitals, which was loaded from NAdO.mwfn.
I haven't tested above procedure yet, but in principle it should work.
Best regards,
Tian
Offline
#9 2022-08-26 15:53:39
- Aerael
- Member
- From: Germany, Bavaria, Munich
- Registered: 2020-10-08
- Posts: 16
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Tian,
thanks so much! ![]()
That should indeed work!
I just an hour ago also had success with a similar method:
I created the basins, exported the AOM, then exported the NAdOs from this AOM, and loaded NAdO.mwfn like suggested by the program automatically. I then used the grid data stored in memory to regenerate the correct basins of the previous SCF density, and subsequently did the orbcomp analysis on the NAdOs within the SCF density basins.
I might switch to your method though, as it allows me to separate AOM creation from orbcomp exporting, which are both pretty time consuming for some of the systems' sizes. I'll have to look wether the .cub files get too big -- those get pretty large sometimes. ![]()
Thank you so, SO much for your continuous support, Tian!
Can we do anything for you as a community besides citing the papers of your work?
Daniel, PhD student at LMU Munich, Germany under Prof. Dr. Klüfers. Special interest: transition metal nitrosyl complexes.
Offline
#11 2022-09-16 13:03:33
- Aerael
- Member
- From: Germany, Bavaria, Munich
- Registered: 2020-10-08
- Posts: 16
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Tian,
unfortunately, there is another closely related issue that I do not understand.
For systems containing heavier elements (like our 3d transition metal complexes), the full real space DI (obtained during the NAdO generation), and the DI from integrating all NAdOs within ALL (!) basins do not match up.
The NAdO integrals rarely overshoot (still, sometimes it happens!), mostly they severely undershoot (my personal record was about 0.8e). Keep in mind that this happens although all atoms of the system are included in the NAdO generation and integration.
This is probably some sort of accuracy issue, as it only happens with post second row elements. I am using a lunatic-quality AOM with mixed grid. I also tried a 0.02 grid spacing, but it gave the same numbers as lunatic (which is comforting). I also tried increasing the box size because I had this issue with some anionic structures, but increasing box size to double the length did not change the numbers significantly either (in the 0.005e range).
So whatever is happening is not due to box size and grid spacing. I could not control anything about the uniform/atomic grid mixture. I do not know if it possible that the error is in there.
This is my echoed multiwfn input string:
17\n1\n1\n4\n6\n2\n-10\n200\n20\n3\n\nATOMS_OF_FRAGMENT_ONE\nATOMS_OF_FRAGMENT_TWO\ny\n0\n17\n1\n2\n11\n-4\n0\n-10\n100\n2\n7\n\n0\nq\n
Thanks again for having another look.
As always, I can send you an example via email.
I hope you have a great week! ![]()
Daniel
Daniel, PhD student at LMU Munich, Germany under Prof. Dr. Klüfers. Special interest: transition metal nitrosyl complexes.
Offline
#12 2022-09-17 19:42:58
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Daniel,
Please send me your inputted wavefunction file (as well as corresponding input file of quantum chemistry program) via E-mail, and show me all commands you inputted in Multiwfn, so that I can exactly reproduce the notable discrepancy you mentioned, then I will check.
Best,
Tian
Offline
#13 2022-09-20 11:22:30
- Aerael
- Member
- From: Germany, Bavaria, Munich
- Registered: 2020-10-08
- Posts: 16
Re: Bug in non-singlet NAdO orbcomp analysis
Dear Tian,
nevermind, it was an accounting error in my code which multiplies the NAdO occs with the orbcomp numbers.
I accessed the wrong array. which only differed by one letter. Classic. *sigh*
Thanks so much nevertheless! ![]()
Daniel
Daniel, PhD student at LMU Munich, Germany under Prof. Dr. Klüfers. Special interest: transition metal nitrosyl complexes.
Offline