Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2022-03-12 05:32:01

QTAIM topology analysis for excited state calculated by CP2K
Today a people asked me how to perform QTAIM topology analysis for excited state calculated by CP2K via Multiwfn, my reply is pasted below, which may be also useful for other users.
The files mentioned in my reply can be downloaded here: diamond_222.rar
---------------------------------
I just updated Multiwfn on its website, the latest version now supports AIM topology analysis for excited state calculated by TDDFT of CP2K.
Diamond 2*2*2 supercell is used to illustrate this point. The TDDFT input file is attached. Note that this task asks CP2K to output .molden file containing molecular orbitals, and "MIN_AMPLITUDE 0.001" option is used to print more configuration state coefficients than default. "ADDED_MOS 100" is used to ask CP2K to solve and save large enough number of virtual orbitals into the .molden file. Also note that using MOLOPT series of basis set is deprecated for present purpose, its cost in Multiwfn for topology analysis is significantly higher than using other basis set with comparable quality. Generally, using TZVP-GTH or TZV2P-GTH is a good choice.
After running the input file, you will obtain .molden file and output file, they are all attached. Note that [ Cell ] and [ Nval ] fields are manually inserted into the .molden file, see Section 2.9.3 of Multiwfn manual for details.
Next, we need to generate a wavefunction file containing natural orbitals of the excited state of interest. Boot up Multiwfn and input
diamond_222.molden
18 // Electron excitation analysis
13 // Generate natural orbitals of excited states
diamond_222.out
1 // The excited state of interest is assumed to be the first excited state[Press ENTER button] // Use 0.25 Bohr of grid spacing to generate overlap matrix(no longer needed in latest version)
After calculation, you will find NO_0001.mwfn has been generated in current folder. Also, from screen you can find this information:
Involved highest unoccupied orbital: 185 (HOMO+ 57)
That means the lowest 57 virtural orbitals are involved in the calculation, therefore setting ADDED_MOS to 100 is indeed large enough.
Then, reboot Multiwfn and use the NO_0001.mwfn as input, after that you can perform AIM topology analysis via main function 2 as usual, see Section 4.2 of Multiwfn manual for example of topology analysis. There is no any difference between the analysis for isolated system and periodic system.
Offline
#2 2022-03-24 13:50:19
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Prefessor
Many thanks for this information. I can calculate an organic molecule with multiwfn. In line with the information you have given above, will I have periodically made a PBC calculation using the calculations in section 4.2 of the NO_0001.mwfn file that I obtained with cp2k (using PBC=periyodic boundry condidation)?
Best Regards
Offline
#3 2022-03-25 00:19:54
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Prefessor
Many thanks for this information. I can calculate an organic molecule with multiwfn. In line with the information you have given above, will I have periodically made a PBC calculation using the calculations in section 4.2 of the NO_0001.mwfn file that I obtained with cp2k (using PBC=periyodic boundry condidation)?
Best Regards
I don't well understand your problem, please further clarify.
Best,
Tian
Offline
#4 2022-03-25 08:28:28
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor, thank you for reply
''Next, we need to generate a wavefunction file containing natural orbitals of the excited state of interest. Boot up Multiwfn and input
diamond_222.molden
18 // Electron excitation analysis
13 // Generate natural orbitals of excited states
diamond_222.out
1 // The excited state of interest is assumed to be the first excited state
[Press ENTER button] // Use 0.25 Bohr of grid spacing to generate overlap matrix''
Is the cp obtained by this method valid for a periodic system or for an isolated molecule in the gas phase?
Best regards
Last edited by taha55 (2022-03-25 08:29:52)
Offline
#5 2022-03-25 09:15:29
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor, thank you for reply
''Next, we need to generate a wavefunction file containing natural orbitals of the excited state of interest. Boot up Multiwfn and input
diamond_222.molden
18 // Electron excitation analysis
13 // Generate natural orbitals of excited states
diamond_222.out
1 // The excited state of interest is assumed to be the first excited state
[Press ENTER button] // Use 0.25 Bohr of grid spacing to generate overlap matrix''Is the cp obtained by this method valid for a periodic system or for an isolated molecule in the gas phase?
Best regards
The process you mentioned above works for all kinds of systems.
By the way, today I just updated Multiwfn on official site, now the step "[Press ENTER button] // Use 0.25 Bohr of grid spacing to generate overlap matrix" is no longer needed, new version computes the matrix analytically, which is much faster and more accurate.
Best,
Tian
Offline
#6 2022-03-25 18:30:47
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor
I will calculate the aim topology, that is, BCP (bond critic point), using the ''diamond_222.molden'' file as in Section 4.2. Is there any need to convert the "diamond_222.molden" file to the "NO_0001.mwfn'' file for this? so we can't use diamond_222.molden file for BCP (bond critic point) calculation as Section 4.2?
Best Regards
Offline
#7 2022-03-26 00:12:44
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor
I will calculate the aim topology, that is, BCP (bond critic point), using the ''diamond_222.molden'' file as in Section 4.2. Is there any need to convert the "diamond_222.molden" file to the "NO_0001.mwfn'' file for this? so we can't use diamond_222.molden file for BCP (bond critic point) calculation as Section 4.2?
Best Regards
The diamond_222.molden mentioned in #1 corresponds to ground state wavefunction. If you simply want to perform topology analysis for ground state, evidently this file can be directly used as input file. If you need to study excited state wavefunction, you have to generate the .mwfn containing excited state natural orbitals via the steps mentioned in #1.
Best,
Tian
Offline
#8 2022-03-26 11:48:22
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor
I calculated cp for vmd using the diamond_222.molden file and as in your video (https://www.youtube.com/watch?v=mgsnhvWH5SI), but the vmd opened molecule was not periodic. I chose the x, y and z coordinates, but the periodic molecules did not form.
Best regards

Last edited by taha55 (2022-03-26 11:50:54)
Offline
#10 2022-03-26 15:09:45
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor,
Thank you very much for patiently answering all my questions. I did what you specified. but only the atoms repeated periodically, the cp did not repeat. Does this indicate that cp's are not calculated in the periodic system?
Best Regards

Last edited by taha55 (2022-03-26 15:13:27)
Offline
#11 2022-03-27 01:11:32
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear taha55,
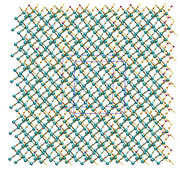
I just updated Multiwfn on official website, now crystal information is also written to paths.pdb and CPs.pdb, thus now you can display their images as molecular structure.
Example ( (3,+3) and (3,+1) are not shown):
Offline
#12 2022-03-27 11:18:09
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor;
You contribute a lot to science and scientists. Really sincerely thank you very much. I want to ask one last thing about this subject. My molden file has 2 atoms Oxygen and Hydrogen. Could you provide some information on what is the [Nval] specified in the multiwfn manual. Also, are the numbers next to the O and H atoms (6 for O and 1 for H) the number of valence electrons?And did I write these numbers correctly?
my file
[Molden Format]
[Cell]
13.0500000 0.00000000 0.00000000
0.00000000 13.0500000 0.00000000
0.00000000 0.00000000 13.0500000
[Nval]
O 6
H 1
[Atoms] AU
O 1 8 0.000000 0.000000 5.680649
H 2 1 -1.609816 0.155501 6.775287
H 3 1 -0.042463 1.710742 3.727409
.
.
.
Last edited by taha55 (2022-03-27 11:47:45)
Offline
#13 2022-03-27 12:05:57
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear taha55,
There is no general answer, the correct values in [Nval] fully depend on the (pseudopotential) basis set you used.
If you are performing all-electron calculation, you do not need to set [Nval].
Assume that you are using MOLOPT series of pseudopotential basis set, you should look into BASIS_MOLOPT file in CP2K. As you can see, there are several basis sets available for oxygen and hydrogen:
H SZV-MOLOPT-GTH SZV-MOLOPT-GTH-q1
H DZVP-MOLOPT-GTH DZVP-MOLOPT-GTH-q1
H TZVP-MOLOPT-GTH TZVP-MOLOPT-GTH-q1
H TZV2P-MOLOPT-GTH TZV2P-MOLOPT-GTH-q1
H TZV2PX-MOLOPT-GTH TZV2PX-MOLOPT-GTH-q1
O SZV-MOLOPT-GTH SZV-MOLOPT-GTH-q6
O DZVP-MOLOPT-GTH DZVP-MOLOPT-GTH-q6
O TZVP-MOLOPT-GTH TZVP-MOLOPT-GTH-q6
O TZV2P-MOLOPT-GTH TZV2P-MOLOPT-GTH-q6
O TZV2PX-MOLOPT-GTH TZV2PX-MOLOPT-GTH-q6
The value after q- is number of valence electrons represented by the basis sets. From above basis set name, it is clear that if you use them in calculation, numbers of valence electron of hydrogen and oxygen are 1 and 6, respectively. So, in this case you should write
[Nval]
O 6
H 1
For some elements, there are large-core pseudopotential and small-core pseudopotential, corresponding to different number of valence electrons; in that case, you should properly write [Nval] according to the actual version of pseudopotential basis set you used.
Offline
#14 2022-03-27 12:35:20
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor
Thank you very much for your help and explanation
Best Regards
Offline
#15 2022-06-11 11:31:55
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor
my file
[Molden Format]
[Cell]
13.0500000 0.00000000 0.00000000
0.00000000 13.0500000 0.00000000
0.00000000 0.00000000 13.0500000
The unit cell lengths of the cubic structure were entered as above. But if the structure is not cubic, How to enter the angle values of the unit cell for a monoclinic structure(α=γ=90°≠β)?
Best Regards
Offline
#16 2022-06-11 12:58:25
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor
my file
[Molden Format]
[Cell]
13.0500000 0.00000000 0.00000000
0.00000000 13.0500000 0.00000000
0.00000000 0.00000000 13.0500000
The unit cell lengths of the cubic structure were entered as above. But if the structure is not cubic, How to enter the angle values of the unit cell for a monoclinic structure(α=γ=90°≠β)?Best Regards
Please note that the three lines define cell vectors, rather than cell lengths and angles. Using cell vectors you can represent any form of cell.
Offline
#17 2022-06-11 13:54:26
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Can you help me on how to do this? how to get lattice vectors?
α=γ=90°, β=103°
a=21 b=11 and c=19 angstrom
Last edited by taha55 (2022-06-11 13:55:13)
Offline
#18 2022-06-12 03:29:55
Re: QTAIM topology analysis for excited state calculated by CP2K
For convenience of use, I update Multiwfn on website today, now you can directly specify cell angles and lengths in .molden file. For your situation, simply add following content into .molden file:
[Cell]
21 11 19 90 103 90Offline
#19 2022-06-12 22:19:26
- taha55
- Member
- Registered: 2019-09-19
- Posts: 38
Re: QTAIM topology analysis for excited state calculated by CP2K
Dear Professor
Thank you very much for your help and update
Best Regards
Offline