Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2022-04-20 05:57:05
- julieborah66
- Member
- Registered: 2018-09-17
- Posts: 32
Intel MKL error: Parameter 8 was incorrect on entry to DGEMM
Dear Sir,
I have a molecule ( consisting of 104 atoms).
I want to calculate the RMSE and RRMSE ( ESP error). I have selected a file containing charges ( RESP Charges) the .chg file and loaded the option 7 Population analysis and atomic charges
Then I went to the 13 Merz-Kollmann (MK) ESP fitting atomic charge.
Again selected the .chg file and then I set the number and scale factors of layers of MK fitting points (1.67 to 2.20)
I selected the 49th atom
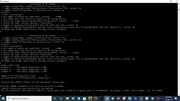
Then this is the error I got
Intel MKL error: Parameter 8 was incorrect on entry to DGEMM and then the Multiwfn software stopped abruptly.
A picture has been attached for reference (P 2. png). With the rest of the atoms, it was giving me values but with two atoms I am getting this error! Also, I would like to add that I have done similar RMSE and RRMSE analyses of CM5 and PCM-CM5 atomic charges, it was working fine!
I did the same thing on my Desktop ( to check if this is a core issue) and then I got NAN issue ( the reference photo: 49 atom problem.png)
Please, help me to sort out the problem!

Offline
#2 2022-04-20 10:33:04

Re: Intel MKL error: Parameter 8 was incorrect on entry to DGEMM
Please paste all commands you inputted in Multiwfn since Multiwfn boots up.
I suspect that you used wrong input file or inputted incorrect commands.
Offline