Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2021-07-14 19:23:04
- mbergami
- Member
- Registered: 2020-01-12
- Posts: 11
Problem with 3.200.6 function
Hi all,
I am getting a strange result with function 3.200.6 when I perform the overlap calculation between the densities (density1 and density2) from some orbitals obtained with two different schemes (case1 and case2).
The radial profiles of the densities from cases 1 and 2 indicate that the overlap should be greater in case 2, but I find a higher result in case 1.
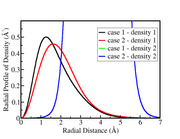
The .wfn files used to calculate the overlaps were taken from converting .molden files.
The wave functions used in these calculations were obtained with ghost centers positioned in different positions in cases 1 and 2. In case 2, more ghost centers are used than in case 1.
I don't understand if the .wfn files are wrong, or if there is some other problem. Can the use of ghost centers affect the density overlap calculation obtained with function 3.200.6?
Thanks for the attention
Mateus
Last edited by mbergami (2021-07-14 19:36:58)
Offline
#2 2021-07-15 08:26:01

Re: Problem with 3.200.6 function
Dear Mateus,
Your representation is somewhat too abstract, I cannot exactly understand the situation you encountered. If possible, please compress your inputted wavefunction files (as well as the input file of quantum chemistry code used to generate them) and then upload here (or send to my E-mail), and show me all commands you inputted in Multiwfn, I will have a check. I need to reproduce your map before answering your question.
Best regards,
Tian
Offline