Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 2021-05-15 19:23:26
- Hans
- Member
- Registered: 2018-10-24
- Posts: 10
Tcl script in VMD
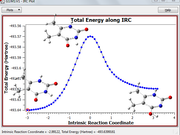
Using a VMD script written by me (IGMinter_isoall.tcl) I try to visualize the behavior of dginter in the course of a reaction (Fig. 1), but the VMD script does not work correctly. Could you check the IGMinter_isoall.tcl script and correct it?
I wrote the IGMinter_isoall.tcl script according to what is indicated in section 4.20.10.2 of the Multiwfn Manual
In RG I attach the files if necessary:
https://www.researchgate.net/post/Tcl_script_in_VMD
---IGMinter_isoall.tcl------------------------------------------------------------------------------------------------
set nsystem 51
set isovaldginter 0.01
set colordginter 7
set colorlow -5.0
set colorhigh 5.0
for {set i 1} {$i<=$nsystem} {incr i} {
set name SP[format %04d $i]
puts "Processing atmdg_$name.pdb and dg_inter_$name.cub..."
display projection Orthographic
display rendermode GLSL
display depthcue off
light 2 on
light 3 on
color Display Background white
axes location Off
color scale method BWR
mol default style CPK
mol new atmdg_$name.pdb
mol new dg_inter_$name.cub
mol modstyle 0 top CPK 1.0 0.8 18.0 16.0
mol modcolor 0 top Beta
mol modmaterial 0 top EdgyShiny
mol scaleminmax 0 top $colorlow $colorhigh
mol modstyle 0 1 Isosurface $isovaldginter 0 0 0 1 1
mol modcolor 0 1 ColorID $colordginter
mol modmaterial 0 1 EdgyGlass
render snapshot $name.bmp
mol delete top
mol delete top
}
Last edited by Hans (2021-05-15 19:24:12)
Offline
#2 2021-06-10 10:02:08
- snljty
- Member
- Registered: 2021-06-10
- Posts: 1
Re: Tcl script in VMD
1. In the first loop, the pdb file is molecule 0 and the cub file is molecule 1. As "top" represents the top molecule, in this loop it is 1, so first, in the first loop you need to change all "top" to "0" to use information in pdb file.
2. In the second loop, although you deleted the first two molecules before, the counter will continue, and so this time the pdb file is molecule 2 and the cub file is molecule 3, and so for other loops.
3. "mol scaleminmax" has a different syntax as other common commands, as the molecule id need to be in front of representation id, and hence it should be "mol scaleminmax top 0" instead of "mol scaleminmax 0 top".
4. Simply there are two solutions: the first is to set a variable as the index of the current loop, and use expression based on this variable to set molecule indices in each loop, and the second is load the cub file INTO the pdb file, and hence there is always one molecule from the beginning to the end, so you can always use "top" to indicate the current molecule. I would change the script to:
set nsystem [llength [glob -nocomplain atmdg_*.pdb]]
set isovaldginter 0.01
set colordginter 7
set colorlow -5.0
set colorhigh 5.0
display projection Orthographic
display rendermode GLSL
display depthcue off
light 2 on
light 3 on
color Display Background white
axes location Off
color scale method BWR
mol default style CPK
for {set i 1} {$i<=$nsystem} {incr i} {
set name SP[format %04d $i]
puts "Processing atmdg_$name.pdb and dg_inter_$name.cub..."
mol new atmdg_$name.pdb
mol addfile dg_inter_$name.cub
animate delete beg 1 end 1 top
animate goto end
mol modstyle 0 top CPK 1.0 0.3 18.0 16.0
mol modcolor 0 top Beta
mol modmaterial 0 top EdgyShiny
mol scaleminmax top 0 $colorlow $colorhigh
mol addrep top
mol modstyle 1 top Isosurface $isovaldginter 0 0 0 1 1
mol modcolor 1 top ColorID $colordginter
mol modmaterial 1 top EdgyGlass
render snapshot $name.bmp
mol delete top
}
Those color Display, axes, color scale ... commands can be moved outside of the loop since they are only needed to be executed once.
p.s.
1. How did you define the fragments? I guess the separation of carbon and oxygen and regard them as "two molecules" is sort of meaningless.
2. Why draw it with such thick bonds (0.8)? Generally 0.2 to 0.3 is most commonly used.
Last edited by snljty (2021-06-10 10:10:31)
Offline
#3 2021-06-10 16:53:39
- Louischarlc0
- Member
- Registered: 2020-07-21
- Posts: 65
Re: Tcl script in VMD
Hi there
Is it possible to obtain the fluorescence lifetime through TD-DFT methods in multiwfn?
Thanks in advance
Regards
Louis-Charl
Offline
#4 2021-06-11 14:48:15

Re: Tcl script in VMD
Hi there
Is it possible to obtain the fluorescence lifetime through TD-DFT methods in multiwfn?
Thanks in advance
Regards
Louis-Charl
This is irrelevant to Multiwfn
You should use Einstein equation based on oscillator strength and excitation energy to estimate lifetime of excited state. You can manually do this very easily, just an algebraic operation.
Offline
#5 2021-06-12 10:08:23
- Louischarlc0
- Member
- Registered: 2020-07-21
- Posts: 65
Re: Tcl script in VMD
thank you so much. I'll try it manually
Regards
Louis-Charl
Offline
Pages: 1