Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 2020-02-23 18:46:11
- rezabma
- Member
- Registered: 2020-02-23
- Posts: 30
ESP for periodic cube
Hi
I have generated a periodic cube file from FHI-aims code that contains ESP data (periodic graphene doped by Al atom). The cube is opened by multiwfn (and also GaussView), but displays NaN for minima and maxima ESP.
Any help.
Last edited by rezabma (2020-03-01 16:17:40)
Offline
#2 2020-02-23 20:58:38

Re: ESP for periodic cube
I don't have FHI-aims code and didn't test compatibility with cube file generated by it. Please detailedly describe your operational steps in Multiwfn (including all commands you inputted, and it is better to paste the output information containing the "NaN" so that I can understand what you refer to)
Offline
#3 2020-02-29 07:28:26
- rezabma
- Member
- Registered: 2020-02-23
- Posts: 30
Re: ESP for periodic cube
Hi, Thanks for your rapid reply. I did following steps:
1- Optimization of the structure by FHI-aims
2- Generating cube file with "output cube hartree_potential" in FHI-aims (ESP cube)
3- reading the cube by Gaussview to check it is correct.
4- opening with multiwfn
5- going to 12
6- then to 0 (start analysis now)
7- getting NaN in "summary of surface analysis": see the picture.
I could upload the cube to test it if there is a place for uploading file (or email you)
Thanks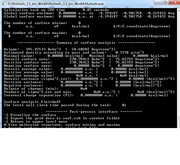
Offline
#4 2020-02-29 07:29:43
- rezabma
- Member
- Registered: 2020-02-23
- Posts: 30
Re: ESP for periodic cube
Please remember that the FHI-aims code is based on NAO (numeric atomic orbitals) similar to Dmol3.
Offline
#5 2020-02-29 07:33:00
- rezabma
- Member
- Registered: 2020-02-23
- Posts: 30
Re: ESP for periodic cube
and this is a simple example (Acetylene cube) generated by FHI-aims and opened by GaussView. As you see there is not any number above it.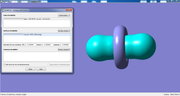
Last edited by rezabma (2020-03-01 16:19:51)
Offline
#6 2020-02-29 23:17:16
Re: ESP for periodic cube
Please note that in order to perform electrostatic potential analysis on molecular surface analysis, commonly you should use a file containing wavefunction information as input file, such as mwfn/wfn/wfx/molden/fch/gms formats, see Section 2.6 of manual for detail, this is because Multiwfn calculates electron density and ESP based on loaded wavefunction during this kind of analysis. Evidently, this analysis cannot be realized solely based on a cube file recording ESP.
Despite FHI-aims is unable to yield a wavefunction file that compatible with Multiwfn, this analysis is still possible if you can provide cube file of both ESP and electron density for your system. In this case, the command for performing the ESP analysis on molecular vdW surface is
density.cub // Load cube file of electron density
12 // Quantiative molecular surface analysis
1 // Select the way to define surface
11 // Isosurface of the grid data in memory
0.001 // Use rho=0.001 a.u. to define the surface
2 // Select mapped function
1 // ESP
5
3 // The mapped function will be interpolated from an external cube file
0 // Start calculation
ESP.cub // The cube file recording ESP
Note that the grid setting used for yielding density.cub and ESP.cub must be exactly the same.
Offline
#7 2020-03-01 07:44:58
- rezabma
- Member
- Registered: 2020-02-23
- Posts: 30
Re: ESP for periodic cube
OK. Thanks. I will test.
Offline
#8 2020-03-01 16:16:58
- rezabma
- Member
- Registered: 2020-02-23
- Posts: 30
Re: ESP for periodic cube
Hi, Thanks for your efforts. I tested above method for my system (periodic Al-doped Graphene), and could generate the pdb file to enter in VMD. However, as you see in the attached file, the origin shifted. In FHI-aims, the origin for my system has been defined in:
cube origin 7.5089 4.3246 9.9289
cube edge 100 0.1500 -0.0868 -0.0012
cube edge 100 0.0002 0.1733 -0.0002
cube edge 100 0.0000 0.0000 0.2000
which shows the origin is x=7.5089, y=4.3246, and z=9.9289.
But seems that Multiwfn automatically choose (0,0,0) for the origin. I have two problems:
1- setting the origin in (7.5089, 4.3246, 9.9289)
2- how to remove layer in the z=20 of the cell (upper layer).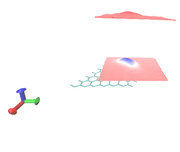
Offline
#9 2020-03-01 19:29:34
Re: ESP for periodic cube
I think the reason is that currently Multiwfn only supports rectangle grid, while the grid in your cube file is evidently not rectangular. The only workaround is properly building a supercell and transform the cell to rectangle type, then use FHI-aims to perform calculation and generate cube files. (Note that Multiwfn never automatically moves origin to 0,0,0)
Currently Multiwfn is mainly designed for analyzing isolated system, there is no option that can remove the upper layer. You should manually erase it using image editor.
Offline
#10 2020-03-02 06:18:00
- rezabma
- Member
- Registered: 2020-02-23
- Posts: 30
Re: ESP for periodic cube
Very thanks. I hope that in the future Multiwfn could read and manipulate periodic jobs. My another question is that when I use density.cube and esp.cube (according to the above method), the summary analysis in Multiwfn says that positive surface area is 0.000 and positive average value is also NaN.
My charge data in FHI-aims show that in Al-doped graphene, Al has positive charge and carbons have near to zero but negative charges. In VMD, the color scale bar shows that Al has negative ESP, and C atoms have positive ESP. I have attached both figures. I think if this problem is solved, I could use the ESP figures in the paper.
Very thanks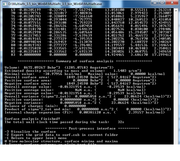
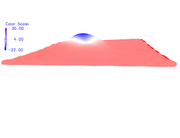
Offline
#11 2020-03-03 02:18:44
Re: ESP for periodic cube
Your Multiwfn version is too old. Please update to the latest version first, then if this problem still exists, please compress the two cube files and then send it to my mail box (the address can be found in the first page of Multiwfn manual)
Offline
#12 2020-04-27 03:17:48
- Yumin Qian
- Member
- Registered: 2020-04-27
- Posts: 7
Re: ESP for periodic cube
Hi
I also tried this method by generation charge density and potential cube file from VASP calculation by files format conversion. And when I followed your suggestion and Multiwfn just stopped at: Loaded CHG.cube successfully! I used the Multiwfn version 3.6 and The cell in my calculation is cubic cell. I also checked the CHG.cube and POT.cube by Vesta and they all show normally. I do not know what happens and can you give me some tips?
Offline
#13 2020-04-27 05:13:48
Re: ESP for periodic cube
Hi
I also tried this method by generation charge density and potential cube file from VASP calculation by files format conversion. And when I followed your suggestion and Multiwfn just stopped at: Loaded CHG.cube successfully! I used the Multiwfn version 3.6 and The cell in my calculation is cubic cell. I also checked the CHG.cube and POT.cube by Vesta and they all show normally. I do not know what happens and can you give me some tips?
I didn't test compatibility with cube file of VASP. Since some codes related to grid data reading has been changed during development of Multiwfn 3.7(dev), I suggest you try 3.7(dev) first, if the latest version is still unable to load your cube file, please upload the cube file to a netdisk and show me link, I will download and try to figure out the reason.
Offline
#14 2020-04-27 09:18:25
- Yumin Qian
- Member
- Registered: 2020-04-27
- Posts: 7
Re: ESP for periodic cube
Dear Sobereva
Thank for your reply and suggestions.
I tried as your suggestion with Multiwfn 3.7, But it remained the same. Here are the files of CHG.cube and POT.cube
https://pan.baidu.com/s/1QgmZluAKMfeg2vD1duVzWg ysdk
Offline
#15 2020-04-27 20:57:13
Re: ESP for periodic cube
I can normally load your cube file using latest version of Multiwfn, as you can see from below output information, the loading is successful.
Input file path, for example E:\iDOLM@STER\Makoto_Kikuchi.wfn
(Supported: .mwfn/wfn/wfx/fch/molden/31/chg/pdb/xyz/mol/mol2/cub/grd, etc.)
Hint: Press ENTER button directly can select file in a GUI window. To reload t
e file last time used, simply input the letter "o". Input such as ?miku.fch can
open the miku.fch in the same folder as the file last time used.
H:\POT.cube
Please wait...
Title line of this file:
CUBE FILE CONVERTED FROM VASP format
OUTTER LOOP: X, MIDDLE LOOP: Y, INNER LOOP: Z
Total number of atoms: 196
Translation vector: X Y Z (Bohr)
Vector 1: 0.130001 0.000000 0.000000
Vector 2: 0.000000 0.130001 0.000000
Vector 3: 0.000000 0.000000 0.130001
The range of x is from 0.000000 to 27.950215 Bohr, 216 points
The range of y is from 0.000000 to 27.950215 Bohr, 216 points
The range of z is from 0.000000 to 27.950215 Bohr, 216 points
Total number of grid points: 10077696
This grid data will take up at least 76 MB memory
Loading grid data, please wait...
The minimum value: -0.41183300E+01 at 3.640028 19.890153 21.190163 Bohr
The maximum value: 0.22817000E+00 at 16.380126 0.390003 26.650205 Bohr
Differential element: 0.0021970507 Bohr^3
Summing up positive value in grid file: 1141005.3814225656
After multiplied by differential element: 2506.8466724032
Summing up negative value in grid file: -1141005.3785599838
After multiplied by differential element: -2506.8466661140
Summing up all value in grid file: 0.0028625818
After multiplied by differential element: 0.0000062892
Loaded H:\POT.cube successfully!
Formula: H28 Li7 C56 N7 O42 F42 S14
Molecule weight: 2766.27302
Point group: C1Then the main function menu is shown, you can use proper options to start analysis.
Offline
#16 2020-04-27 21:49:44
- Yumin Qian
- Member
- Registered: 2020-04-27
- Posts: 7
Re: ESP for periodic cube
Did you use the Linux version? I tried the Linux version on my server because my laptop is not strong enough.
Here is the output information
Differential element: 0.0021970507 Bohr^3
Summing up positive value in grid file: 1141005.3814225656
After multiplied by differential element: 2506.8466724032
Summing up negative value in grid file: -1141005.3785599838
After multiplied by differential element: -2506.8466661140
Summing up all value in grid file: 0.0028625818
After multiplied by differential element: 0.0000062892
Loaded POT.cube successfully!
^Cforrtl: error (69): process interrupted (SIGINT)
Image PC Routine Line Source
Multiwfn 0000000001AF4D64 Unknown Unknown Unknown
libpthread-2.17.s 00002B10BA0C05F0 Unknown Unknown Unknown
libc-2.17.so 00002B10BA3C0BED __poll Unknown Unknown
libxcb.so.1.1.0 00002B10BBBB305A Unknown Unknown Unknown
libxcb.so.1.1.0 00002B10BBBB0E43 xcb_connect_to_fd Unknown Unknown
libxcb.so.1.1.0 00002B10BBBB4A73 xcb_connect_to_di Unknown Unknown
libX11.so.6.3.0 00002B10B9826F0A _XConnectXCB Unknown Unknown
libX11.so.6.3.0 00002B10B9817D92 XOpenDisplay Unknown Unknown
Multiwfn 0000000000B81EAC Unknown Unknown Unknown
Multiwfn 0000000000B9F634 Unknown Unknown Unknown
Multiwfn 00000000007934FB Unknown Unknown Unknown
Multiwfn 0000000000430922 Unknown Unknown Unknown
libc-2.17.so 00002B10BA2EF505 __libc_start_main Unknown Unknown
Multiwfn 0000000000430829 Unknown Unknown Unknown
Offline
#17 2020-04-27 23:02:05
Re: ESP for periodic cube
On my computer, both Windows and Linux version of Multiwfn can normally load your file.
This problem of Linux version commonly implies that the Multiwfn has not been installed properly, please carefully follow the installation steps in Section 2.1.2 of Multiwfn maual.
Offline
#18 2020-04-28 06:42:53
- Yumin Qian
- Member
- Registered: 2020-04-27
- Posts: 7
Re: ESP for periodic cube
Dear Lu
I followed your suggestion and check each item for installing multiwfn and it remained the same.
So, I find a new computer with win10 and tried the multiwfn_3.7_dev. And the program collapsed at
0 // Start calculation
ESP.cub // The cube file recording ESP
I do not know what the problem. Can you check what happens?
Offline
#19 2020-04-28 19:36:04
Re: ESP for periodic cube
Dear Lu
I followed your suggestion and check each item for installing multiwfn and it remained the same.
So, I find a new computer with win10 and tried the multiwfn_3.7_dev. And the program collapsed at
0 // Start calculation
ESP.cub // The cube file recording ESP
I do not know what the problem. Can you check what happens?
I updated Multiwfn today, this version is able to normally perform quantitative molecular surface analysis based on your cube file, the resulting map is shown below
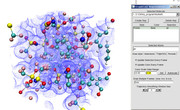
If the grid data contained in the POT.cub is ESP, the map seems to be weird. Since I am familiar with VASP, I cannot give you more advice.
Offline
#20 2020-04-29 04:47:55
- Yumin Qian
- Member
- Registered: 2020-04-27
- Posts: 7
Re: ESP for periodic cube
I updated Multiwfn today, this version is able to normally perform quantitative molecular surface analysis based on your cube file, the resulting map is shown below
Thank you very much for your reply. I am sure there is no problem of charge and potential. CHG.cube is the charge density and POT.cube is the Local Hartree potential from LOCPOT output file. I think the abnormal may comes from the mismatch of VASP and Multiwfn. I will check more carefully.
Thank you again for your suggestion and patience
Offline
#21 2020-04-29 05:52:20
Re: ESP for periodic cube
Hartree potential is different to ESP, it only corresponds to the electrostatic potential due to electron density. In contrast, in the definiton of ESP, the nuclear charge contribution is also taken into account.
Offline
#22 2020-04-29 07:12:54
- Yumin Qian
- Member
- Registered: 2020-04-27
- Posts: 7
Re: ESP for periodic cube
Hartree potential is different to ESP, it only corresponds to the electrostatic potential due to electron density. In contrast, in the definiton of ESP, the nuclear charge contribution is also taken into account.
So you think I should use the Total electrostatic potential, which also include the XC part ? Because vasp already include the ionic+Hartree in the output file.
Last edited by Yumin Qian (2020-04-29 07:23:26)
Offline
#23 2020-04-29 20:28:11
Re: ESP for periodic cube
You don't need to include the XC potential. If you can ensure that the current POT.cube includes ionic (nuclear contribution) + Hartree (classical electronic contribution), in principle it should directly correspond to ESP (but the sign may need to be inverted since electron carries one unit of negative charge).
Since your system is too complex, I suggest you first using VASP to calculate a simple system such as a water molecule to check whether the ESP over the vdW surface can be normally produced via the aforementioned way.
Offline
Pages: 1