Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2024-04-16 12:40:50
- Quark
- Member
- Registered: 2020-06-24
- Posts: 15
Local integration of electron density
Dear Dr. Tian Lu,
I am trying to study non-covalent interactions between O and S atoms within a single molecule. However, I am encountering an issue with integrating the electron density, as I cannot find the NOCV pair associated solely with this interaction. Consequently, the density of the entire NOCV pair is integrated, leading to an overestimation of the number of participating electrons. The image suggests that there is no electron density between the O and S atoms, yet the CDF curve indicates otherwise. If there is a way to integrate the electron density along the Z direction for selected atoms only, without considering the rest of the electron density of the molecule.
I would be deeply grateful to you for any comments.
--
Best regards,
Eugene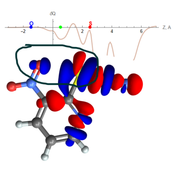
Offline
#2 2024-04-16 17:32:17

Re: Local integration of electron density
Dear Eugene,
You can consider to use BOD/NAdO instead of ETS-NOCV. Performing BOD/NAdO analysis doesn't need definition and individual calculations of fragments, you can just specify two atoms, then covalent interaction between the two atoms can be studied, see Section 4.200.20 of Multiwfn manual for examples. There is no way to exactly confine the ETS-NOCV analysis between the two atoms in your system.
Best,
Tian
Offline
#3 2024-04-17 10:19:19
- Quark
- Member
- Registered: 2020-06-24
- Posts: 15
Re: Local integration of electron density
Dear Dr. Tian Lu,
Thank you very much for confirming my concerns about the ETS-NOCV analysis.
I will try to use the BOD/NAdO need method you proposed to analyze the interaction.
Best regards,
Eugene
Offline