Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 2026-06-29 17:56:08
- b322qr
- Member
- Registered: 2024-04-30
- Posts: 10
Multiwfn AdNDP Analysis with ORCA Output File
I am performing AdNDP analysis on molecules using option 14 in Multiwfn. I am performing the NBO calculation using ORCA and using the .out file as the input to Multiwfn. Everything works fine at this point. I can get the 1-center, 2-center, etc orbitals with what seems like the correct occupation numbers and %contributions from each participating atom. Then, when I want to visualize AdNDP analysis orbitals (using option 9 from the AdNDP submenu) from an NBO output file from an ORCA calculation, I create a .fch file by first making a .molden.input file and then converting it to a .fch file using option 100 then option 2 then option 7 in Multiwfn. However, the visualized orbitals are very weird. They do not look like typical orbitals you would expect. Even the O-H bond in water has an odd shape, which is totally delocalized over the entire molecule, that is not what you would expect from AdNDP analysis. I also have access to Gaussian and have run NBO calculations in Gaussian and performed AdNDP analysis with no problem.
Is there any way to fix this problem with ORCA? Is there a known workaround other than resorting to Gaussian?
Offline
#2 2026-06-30 09:51:38

Re: Multiwfn AdNDP Analysis with ORCA Output File
Perhaps the AONAO matrix (transformation coefficients between basis functions and NAOs) was not properly loaded. The AdNDP function in Multiwfn has not been formally supported for ORCA+NBO combination...
Online
#3 2026-07-01 12:18:54
- b322qr
- Member
- Registered: 2024-04-30
- Posts: 10
Re: Multiwfn AdNDP Analysis with ORCA Output File
Thank you for the response, this was helpful.
I have one more question. Do you expect the composition of the AdNDP orbitals (i.e., for the O-H bond in water: %O composition, %2s/2p composition) to be reliable from an ORCA+NBO output file? I am currently working with an organometallic complex (optimized in ORCA followed by an NBO calculation in ORCA) and need to extract information about the polarization and hybridization of the metal-ligand bond.
Offline
#4 2026-07-02 08:35:08
Re: Multiwfn AdNDP Analysis with ORCA Output File
I think the composition should be correct, but I didn't have a try. You can compare your result with the example in Multiwfn manual to confirm this.
Online
#5 2026-07-07 01:10:59
- b322qr
- Member
- Registered: 2024-04-30
- Posts: 10
Re: Multiwfn AdNDP Analysis with ORCA Output File
I'm posting my reply here for future user with the same question to see the results.
I used the phenanthrene molecule from section 4.14.3 in the Multiwfn manual and compared what I believe is the same calculation in ORCA. For others to see, here is the input file for the Gaussian calculation:
# b3lyp/3-21g pop=nboread
b3lyp/3-21g opted
0 1
C 0.00000000 3.56061700 -0.29722900
C 0.00000000 2.83932500 0.87979300
C 0.00000000 1.42361400 0.86771500
C 0.00000000 0.72986200 -0.38070000
C 0.00000000 1.49924400 -1.56931800
C 0.00000000 2.88151800 -1.53149000
C 0.00000000 0.67926500 2.09717700
C 0.00000000 -0.72986200 -0.38070000
C 0.00000000 -1.42361400 0.86771500
C 0.00000000 -0.67926500 2.09717700
C 0.00000000 -2.83932500 0.87979300
H 0.00000000 -3.34927800 1.83744900
C 0.00000000 -3.56061700 -0.29722900
C 0.00000000 -2.88151800 -1.53149000
C 0.00000000 -1.49924400 -1.56931800
H 0.00000000 1.23356400 3.02968400
H 0.00000000 4.64400700 -0.27588200
H 0.00000000 3.34927800 1.83744900
H 0.00000000 1.00203800 -2.53051300
H 0.00000000 3.44643700 -2.45642700
H 0.00000000 -1.23356400 3.02968400
H 0.00000000 -4.64400700 -0.27588200
H 0.00000000 -3.44643700 -2.45642700
H 0.00000000 -1.00203800 -2.53051300
$nbo dmnao aonao $end
And here is the input for the ORCA calculation:
! B3LYP 3-21G NBO
%nbo
nbokeylist="$nbo aonao dmnao $end"
end
%MaxCore 7500
%pal nprocs 16
end
* xyz 0 1
C 0.00000000 3.56061700 -0.29722900
C 0.00000000 2.83932500 0.87979300
C 0.00000000 1.42361400 0.86771500
C 0.00000000 0.72986200 -0.38070000
C 0.00000000 1.49924400 -1.56931800
C 0.00000000 2.88151800 -1.53149000
C 0.00000000 0.67926500 2.09717700
C 0.00000000 -0.72986200 -0.38070000
C 0.00000000 -1.42361400 0.86771500
C 0.00000000 -0.67926500 2.09717700
C 0.00000000 -2.83932500 0.87979300
H 0.00000000 -3.34927800 1.83744900
C 0.00000000 -3.56061700 -0.29722900
C 0.00000000 -2.88151800 -1.53149000
C 0.00000000 -1.49924400 -1.56931800
H 0.00000000 1.23356400 3.02968400
H 0.00000000 4.64400700 -0.27588200
H 0.00000000 3.34927800 1.83744900
H 0.00000000 1.00203800 -2.53051300
H 0.00000000 3.44643700 -2.45642700
H 0.00000000 -1.23356400 3.02968400
H 0.00000000 -4.64400700 -0.27588200
H 0.00000000 -3.44643700 -2.45642700
H 0.00000000 -1.00203800 -2.53051300
*
In the Multiwfn manual, the residual density distribution after accepting the first 27 orbitals is:
1 C : 1.0250 2C : 1.0370 3C : 1.0280 4C : 1.0414
5 C : 1.0339 6C : 1.0262 7C : 0.1322 8C : 1.0414
9 C : 1.0280 10C : 0.1322 11C : 1.0370 12H : 0.0117
13C : 1.0250 14C : 1.0262 15C : 1.0339 16H : 0.0121
17H : 0.0113 18H : 0.0117 19H : 0.0126 20H : 0.0111
21H : 0.0121 22H : 0.0113 23H : 0.0111 24H : 0.0126
In my ORCA calculations, I receive the following:
1C : 1.0234 2C : 1.0295 3C : 1.0243 4C : 1.0381
5C : 1.0246 6C : 1.0246 7C : 0.1302 8C : 1.0473
9C : 1.0167 10C : 0.1341 11C : 1.0345 12H : 0.0112
13C : 1.0234 14C : 1.0246 15C : 1.0217 16H : 0.0115
17H : 0.0106 18H : 0.0112 19H : 0.0120 20H : 0.0104
21H : 0.0115 22H : 0.0106 23H : 0.0104 24H : 0.0120
Later, the analysis of orbital 31 shows the following in the Multiwfn manual:
NAO# Center Label Type Composition
67 8(C ) px Val( 2p) 2.247%
76 9(C ) px Val( 2p) 1.929%
94 11(C ) px Val( 2p) 13.276%
105 13(C ) px Val( 2p) 32.846%
114 14(C ) px Val( 2p) 34.484%
123 15(C ) px Val( 2p) 15.204%
In my ORCA calculations, I receive the following:
NAO# Center Label Type Composition
67 8(C ) px Val( 2p) 2.217%
76 9(C ) px Val( 2p) 1.942%
94 11(C ) px Val( 2p) 13.387%
105 13(C ) px Val( 2p) 32.956%
114 14(C ) px Val( 2p) 34.401%
123 15(C ) px Val( 2p) 15.082%
As you can see, the differences between the two calculations in not very significant (>0.2 %); however, the visualization of the orbitals is still weird.
These are the orbitals from the Multiwfn manual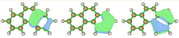
And here are the orbitals from my calculation (using a contour value of 0.03)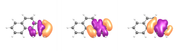
Offline