Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Quantum Chemistry » Convergence criterion and energy accuracy » 2021-03-19 02:26:46
Thanks alot for the advice!
#2 Quantum Chemistry » Convergence criterion and energy accuracy » 2021-03-17 23:06:00
- lmangin
- Replies: 2
Dear Pr. Lu,
I am working on a project with system/basis set that is too heavy for a proper SCF convergence, with 64G memory over 7 days. I tried it with the regular algorithm, with the qc, xqc and yqc algorithms.
In some cases, the normal algorithm has time end up with "convergence criterion not met" (after the default of 129 cycles), however, Gaussian ends up with a "Normal termination".
I noticed that the energy is quite similar and oscillating around a value, for which the difference at each iteration is small (< 0.00000001 Ha) thus doesn't seem significant for an PES of a chemical reaction with an accuracy of 0.1 kcal/mol. However, I am wondering if this electronic energy is actually meaningless since it did not fully converge, or if it could be used as a single point for the electronic single point calculations of the reaction surface.
Thanks in advance!
#3 Re: Quantum Chemistry » Diffuse functions of Hydrogen atoms: s function or p function? » 2021-01-24 23:37:11
Thanks again for your time and your answer. The study is done with B3LYP and M06-2X functionals.
I forgot to mention that in the system, there is a counter anion and some intermediates in the whole process bear fluorine atoms like (F3C-SO2)2N- anion. This is the main reason I had added diffuse functions in Pople's basis set for the geometry optimizaiton and thermochemistry.
You mentioned that def2-TZVP would work better than Pople's and I totally agree but was rather aiming at using Karlruhe's for single points only (so the calculation is less exepensive in time). But now, considering the presence of anion and fluorine atoms, I need to keep diffuse functions, so I guess def2-TZVPD would be more appropriate that def2-TZVP... but again there will only be a 'p' diffuse function for the hydrogens.. that's why I'm wondering if it was reasonable do the single points with def2-TZVPD for non-H and 6-311++G** for H; or if using def2-TZVPD for all atoms (including H) would cause a problem/errors in the electronic energy calculation.
Otherwise, I will read over the pcseg series and consider it for the electronic energies.
Thanks in advance for your input!
#4 Re: Quantum Chemistry » Diffuse functions of Hydrogen atoms: s function or p function? » 2021-01-24 17:49:15
Thank you for you reply. The hydride I'm studying is transferred from a boron to a carbon atom, so it is not a 'free' hydride, and the highest transition state (the H- transfer) has C-H-B atoms quite linear.
The whole energy surface (geometry optimization and thermochemistry) has been calculated with 6-311++G** for the whole molecule bearing 'light atoms' (heaviest is nitrogen), but now I am looking for a higher basis set for single point calculations.
Would it be recommended to use a mixed basis set of def2-TZVPD for all non-hydrogen atoms, and 6-311++G** for hydrogen atoms only ? Or is it best to not do single points for electronic energies with def2 sets at all?
Thanks in advance.
#5 Quantum Chemistry » Diffuse functions of Hydrogen atoms: s function or p function? » 2021-01-24 00:35:44
- lmangin
- Replies: 4
Dear all,
I am studying the mechanism of an organic reaction involving a hydride transfer.
Looking into the various basis sets on BasisSetExchange, I looked into the basis functions and coefficients for hydrogen atoms and am puzzled to notice this:
- the difference between 6-311+G** and 6-311++G** is an 's' function
- the difference between def2-TZVP and def2-TZVPD is a 'p' function
What is the reason for the differences in Pople vs. Karlsruhe basis sets?
Why is the diffuse function an s orbital in one case, and a p orbital in the other ?
How important is that difference and what impact will it have on the accuracy of the calculation?
Thanks in advance!
#6 Re: Multiwfn and wavefunction analysis » Multiwfn ELF calculation » 2020-02-20 13:47:22
I actually realized that I had added with Topmod a wrong symmetry. Maybe this thread should be deleted since my question arises from a mistake and is eventually not relevant to Multiwfn users.
Thank you for you quick reply, and thank you for the Multiwfn package which is really clear and user friendly!
#7 Multiwfn and wavefunction analysis » Multiwfn ELF calculation » 2020-02-19 20:10:07
- lmangin
- Replies: 2
Hi all,
I am a beginner on Electron Localization Function analysis and am practicing on the acetone molecule.
I generated the ELF grid with Topmod and with Multiwfn, using the same .wfn input file.
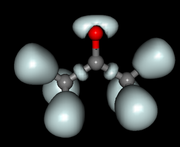
It seems like both softwares give different results wehn I vizualize the cubes with Molekel (see images attached: the surfaces with the 4 large spherical basins around the O atom has been generated with Topmod, and the other one with Topspin.
Is Multiwfn calculating the ELF in a different way? Or did I make a mistake somewhere when generating the grid ?
Thanks in advance.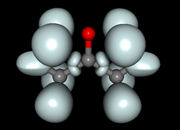
Pages: 1