Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Multiwfn and wavefunction analysis » Electron excitation analysis of a very large wavefunction » 2025-11-02 18:09:35
- mbergami
- Replies: 1
Hi all,
I am trying to perform excitation analysis only for the first excited state of a system with 561 atoms. I employed TD-DFT/6-311G(d,p) with LC-wHPBE using Gaussian 16. Now I have the .log and .fchk of this wavefunction. Moreover, I have 300 wavefunctions of different geometries of this system to analyse in the same way, and I need to accelerate the excitation analysis.
I want to perform the analysis with the function “18 Electron excitation analysis” and “1 Analyze and visualize hole&electron distribution, transition density, and transition electric/magnetic dipole moment density” to get fragment contributions using “2 Hirshfeld partition”.
The system and wavefunction are very large, so it’s too slow. Is there any way to accelerate? Is there an alternative using Multiwfn's other functionalities to perform this analysis faster?
In principle, I need the fragments' contributions for hole and electron only of S1, hole and electron overlap, and, if possible, also perform an IFCT analysis to determine the CT or LE character of this excited state.
Any help will be very welcome!
#2 Multiwfn and wavefunction analysis » Contributions for the HF orbital energy. How could we calculate it? » 2024-06-15 19:01:22
- mbergami
- Replies: 1
Hi everyone
Is it possible to calculate the contributions for a specific HF orbital energy through Multiwfn or Gaussian 16? For example, my system comprises ten water molecules and one extra electron. I want to calculate the contributions of core Hamiltonian, Coulomb integrals, and Exchange integrals for the SOMO energy. I aim to evaluate these three contributions for different cluster sizes.
I can calculate these contributions through a .wfn file containing all orbitals of this wave function or even from a .fchk/.chk file generated in an HF single-point calculation.
Many thanks
Mateus
#3 Multiwfn and wavefunction analysis » Coulomb and four-center integrals with specific orbitals » 2022-08-19 00:30:46
- mbergami
- Replies: 1
Hi all,
I need to calculate Coulomb and four-center integrals between specific orbitals from different molden files.
1- The Coulomb integrals are different from the usual Coulomb integrals between electronic orbitals, in this case the integrals are:
(ii'|jj') = int ( phi_i(r1) phi_i'(r2) r12 phi_j(r1) phi_j'(r_2) )dr1 dr2
the molecular orbitals phi_i(r1) and phi_j(r1) are from the same molden file, while both orbitals phi_i'(r2) and phi_j'(r2) are saved in other molden file.
2- The four-center integrals are a specific case of overlap integrals, in this case the integrals are:
S_(iji'j') = int ( phi_i(r) phi_j(r) phi_i'(r) phi_j'(r) )dr
phi_i and phi_j are molecular orbitals from the same molden file, phi_i' and phi_j' are molecular orbitals from other molden file as in the case of Coulomb integrals.
I am sending in this message also the equations of Coulomb integral (coulomb_integral.jpeg) and four-center integral (four_center_integral.jpeg) attached.
Is there any way to calculate this special cases of coulomb and four-center integrals with Multiwfn code?
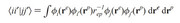
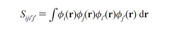
#4 Multiwfn and wavefunction analysis » Problem with 3.200.6 function » 2021-07-14 19:23:04
- mbergami
- Replies: 1
Hi all,
I am getting a strange result with function 3.200.6 when I perform the overlap calculation between the densities (density1 and density2) from some orbitals obtained with two different schemes (case1 and case2).
The radial profiles of the densities from cases 1 and 2 indicate that the overlap should be greater in case 2, but I find a higher result in case 1.
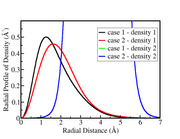
The .wfn files used to calculate the overlaps were taken from converting .molden files.
The wave functions used in these calculations were obtained with ghost centers positioned in different positions in cases 1 and 2. In case 2, more ghost centers are used than in case 1.
I don't understand if the .wfn files are wrong, or if there is some other problem. Can the use of ghost centers affect the density overlap calculation obtained with function 3.200.6?
Thanks for the attention
Mateus
#5 Quantum Chemistry » Fourier transform of molecular orbitals » 2020-06-28 21:44:37
- mbergami
- Replies: 0
Hi all,
Does anyone know how to computationally calculate the Fourier transform of the product of two molecular orbitals? I have molecular orbitals at position space obtained from quantum calculations, and I need the Fourier transform of product of orbitals.
Thanks,
Mateus
#6 Re: Multiwfn and wavefunction analysis » Overlap between specific orbitals » 2020-05-29 21:33:25
Hi Tian, thanks for your help as usual!
I used the subfunction 6 of main function 200 and the result for overlap integrals between square of orbitals are too high, for my system the overlap is 0.91 and the overlap of square is 0.93. I think there is something wrong...
I did other test with two .wfn files of alpha and beta orbitals respectively, and the overlap of square calculated with subfunction 6 of main function 200 is different than the result obtained with the subfunction 11 of main function 100 using just one file with alpha and beta electrons.
#7 Re: Multiwfn and wavefunction analysis » Overlap between specific orbitals » 2020-05-27 16:46:13
Dear Tian,
I am trying to obtain overlap integral of square of the two orbitals from two wavefunctions files. I just know to calculate this overlap integral using function 3.100.11, in this case I can't use two wavefunctions files with this function.
In this case, is there some way to calculate the overlap integral of square of the two orbitals without to merge the two wavefunctions files?
Thanks for the attention,
Mateus Bergami
#8 Re: Multiwfn and wavefunction analysis » Overlap between specific orbitals » 2020-03-19 19:48:43
In this case, I modify the molden file of alpha electrons including the positron orbital and output the .wfn for this modified molden file.
#9 Multiwfn and wavefunction analysis » Overlap between specific orbitals » 2020-03-19 19:40:02
- mbergami
- Replies: 6
Hi all,
I need to calculate the overlap between two specific orbitals obtained with UHF calculation for a system compused by 10 water molecules,
one additional electron and one positron. In this calculation, I positioned GTF's for electrons on atom coordinates, and also a extra expansion center on origin (center of cavity). The GTF's positioned on atom coordinates (6-31G+(d,p)) and on origin (aug-cc-pVTZ) are different.
For the positron I positioned just one expansion center on origin with the same GTF (aug-cc-pVTZ) used for extra center of electrons. Therefore, the electronic wave function has different GTF's (6-31G+(d,p) and aug-cc-pVTZ) than positronic wave function (aug-cc-pVTZ). I have separate molden files for beta electrons, alpha electrons and the positron.
My question is:
Is there some problem to create just one .wfn file for alpha electrons and positron, and use this file to calculate overlap between alpha orbital with positron orbital using the function "3.200.10 Output various kind of integral between orbitals"? Is this function use the information of GTF's to calculate the overlap?
Thanks for the attention,
Mateus Bergami
#10 Multiwfn and wavefunction analysis » Spherically averaged function for spin density » 2020-02-04 14:22:54
- mbergami
- Replies: 1
Hi all,
I would like to know what is the difference between Radial Distribuction Function (RDF) of spin density and spherically averaged spin density.
When I calculate RDF of spin density the program give me the option "7 Export the spherically averaged function", and it isn't clear to me what is this because the RDF function already give me the integral on angular coordinates.
Thanks for the attention,
Mateus Bergami
#11 Multiwfn and wavefunction analysis » How to convert two .molden files (alpha and beta orbitals)? » 2020-01-12 22:14:38
- mbergami
- Replies: 1
Hi everyone,
My question is how could I convert two .molden files from alpha and beta orbitals into just one .wfn file? I know how to convert just one .molden file into a .wfn with Multiwfn program.
I need this .wfn to calculate the spin density of electrons, because actually I have a .molden of alpha electrons and other for beta electrons.
Thanks,
Mateus Bergami
Pages: 1