Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 Multiwfn and wavefunction analysis » How to visualize just the bond critical point and bond path » 2021-09-28 02:03:10
- shenaya
- Replies: 1
Dear All,
I want to generate electron density and Laplacian of electron density maps just with bond critical points and bond paths. Withaout and ring or cage critical points.

I'm doing below steps.
Load the file.
2 topological analysis
2 Search CPs from nuclear positions
3 Search CPS from midpoints of atom pairs
8 generating the paths connecting (3,-3) and (3,-1)
then plot Laplacian of electron density or electron density
But I want to remove ring critical points
Can you please advise me on this
Thanks
Gayani
#2 Re: Multiwfn and wavefunction analysis » Calculating emission spectra » 2021-05-27 00:59:30
Thank you.
#3 Re: Multiwfn and wavefunction analysis » Calculating emission spectra » 2021-05-25 10:41:40
Dear Tian,
This is a part of the output file for fluorescence emission calculation.
Input : #p opt td=(singlets,root=1) b3lyp/genecp scrf=(solvent=methanol) geom=
connectivity
I have calculated emission spectra. When do orbital contribution calculation I have to consider transition from 145 ---->147 ?
or I understood this wrongly?
Excitation energies and oscillator strengths:
Excited State 1: Singlet-A 1.2577 eV 985.84 nm f=0.0047 <S**2>=0.000
145 ->147 0.68259
This state for optimization and/or second-order correction.
Total Energy, E(TD-HF/TD-KS) = -3027.47515828
Copying the excited state density for this state as the 1-particle RhoCI density.
Excited State 2: Singlet-A 1.7817 eV 695.89 nm f=0.0001 <S**2>=0.000
144 ->147 -0.67710
Excited State 3: Singlet-A 1.9683 eV 629.89 nm f=0.0027 <S**2>=0.000
145 ->146 0.69194
SavETr: write IOETrn= 770 NScale= 10 NData= 16 NLR=1 NState= 3 LETran= 64.
The selected state is a singlet
#4 Re: Multiwfn and wavefunction analysis » Calculating emission spectra » 2021-05-14 02:46:41
Hi Tian,
I have successfully plot fluorescence spectra by following the instructions stated by you (http://sobereva.com/wfnbbs/viewtopic.php?id=413).
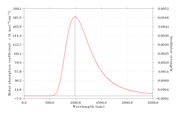
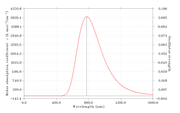
1. My question is can we plot phosphorescence spectra using exactly same steps?
11 // Plotting spectra
3 // UV-Vis
20 // Modify oscillator strengths
2-3 // Choose S2 and S3 states
0 // Set oscillator strengths to zero (because emission is from S1 state)
0 // Plot spectrum
Becouse phosphorescence is T1---->S0 transition.
Another thing is when people mean emission spectra which one in transition metal complexes.
fluorescence, phosphorescence, or both?
Thank you
#5 Re: Multiwfn and wavefunction analysis » Calculating emission spectra » 2021-05-11 01:32:04
Thank you very much !!!
#6 Re: Multiwfn and wavefunction analysis » Calculating emission spectra » 2021-05-07 08:29:57
Thank you, Tian. Will do it. Generally how many nstates should be used for this type of transition metal complexes.
#7 Multiwfn and wavefunction analysis » Calculating emission spectra » 2021-05-07 03:56:54
- shenaya
- Replies: 9
Dear Tian,
I need to plot emission spectra of Ru transition metal complexes.
This is my input for optimization.
Is this correct?
I need both fluorescence and phosphorescence.
Can I take the output of this calculation to obtain the emission spectra using Multiwfn
%mem=4GB
%nprocshared=4
%chk=Neutral.chk
#p td=(50-50,nstates=10,root=1) b3lyp/genecp scrf=(solvent=methanol)
guess=save geom=connectivity opt
Neutral Fluoride Complex from the X-ray crystal structure
0 1
Ru 0.49152900 -1.00287600 0.09326700
Cl 0.66506000 -1.18796400 -2.33847900
Cl 0.96242300 -1.06595000 2.50569800
P -1.14911300 0.54149000 0.02331800
F 2.76034900 -1.90860800 0.00631700
N 1.77532400 0.64178100 -0.02843200
O -2.22603700 -2.70087200 0.96397000
S -0.87762300 -2.87042900 0.29049200
C 1.27960300 1.90403300 -0.05083200
C -0.13813900 2.08851300 -0.01943800
C -0.64790200 3.36905700 0.00649500
H -1.72172700 3.52421800 0.05440300
C 0.20504300 4.50762600 -0.02719900
H -0.23475300 5.49948700 -0.01111100
C 1.56632600 4.35252600 -0.08013500
H 2.22538600 5.21520600 -0.10762500
C 2.14873400 3.04896800 -0.08989900
C 3.53085900 2.83652700 -0.12465600
H 4.20665800 3.68669100 -0.15556800
C 4.05139700 1.53821100 -0.11568800
C 5.45120700 1.25536100 -0.15050600
H 6.14675600 2.08773100 -0.19077900
C 5.90357800 -0.03875900 -0.13249900
H 6.96691600 -0.25133600 -0.15894000
C 4.98721500 -1.12783200 -0.07836500
H 5.33654100 -2.15449100 -0.06390000
C 3.65038400 -0.86564600 -0.04418000
C 3.11234700 0.44907000 -0.06204300
C -1.19758300 -3.75212800 -1.26942600
H -1.79914200 -4.63222900 -1.02933400
H -0.25052300 -4.02752200 -1.73610500
H -1.74514100 -3.07082800 -1.92117300
C -0.00292000 -4.20020300 1.17802700
H 0.23376000 -3.81407100 2.17029700
H 0.91641400 -4.44979400 0.64186000
H -0.66776000 -5.06540600 1.23910000
C -2.18596100 0.55229000 -1.50150700
C -3.18113500 -0.43327200 -1.62938500
H -3.34446200 -1.14905100 -0.82796800
C -3.96636500 -0.49594700 -2.78073900
H -4.73480500 -1.25924300 -2.86603300
C -3.76744600 0.42012100 -3.81820500
H -4.38023800 0.37087500 -4.71387100
C -2.78018900 1.39921300 -3.69735000
H -2.62036000 2.11581000 -4.49811900
C -1.99099500 1.46594500 -2.54554700
H -1.22701800 2.23231700 -2.46900500
C -2.38962000 0.87519600 1.35444700
C -3.60606500 1.51256500 1.05151600
H -3.85995700 1.75348300 0.02521400
C -4.50566300 1.84038400 2.06810900
H -5.44108100 2.33239300 1.81668500
C -4.20643400 1.53429500 3.39706000
H -4.90946000 1.78576800 4.18632200
C -3.00006700 0.90154900 3.70558200
H -2.76052400 0.65709200 4.73687400
C -2.09536100 0.57430500 2.69378500
H -1.16252800 0.08069500 2.94054800
1 4 1.0 8 1.0
2
3
4 10 1.0 37 1.0 48 1.0
5 27 1.0
6 9 1.5 28 1.5
7 8 1.5
8 29 1.0 33 1.0
9 10 1.5 17 1.5
10 11 2.0
11 12 1.0 13 1.5
12
13 14 1.0 15 2.0
14
15 16 1.0 17 1.5
16
17 18 1.5
18 19 1.0 20 1.5
19
20 21 1.5 28 1.5
21 22 1.0 23 2.0
22
23 24 1.0 25 1.5
24
25 26 1.0 27 2.0
26
27 28 1.5
28
29 30 1.0 31 1.0 32 1.0
30
31
32
33 34 1.0 35 1.0 36 1.0
34
35
36
37 38 1.5 46 1.5
38 39 1.0 40 1.5
39
40 41 1.0 42 1.5
41
42 43 1.0 44 1.5
43
44 45 1.0 46 1.5
45
46 47 1.0
47
48 49 1.5 57 1.5
49 50 1.0 51 1.5
50
51 52 1.0 53 1.5
52
53 54 1.0 55 1.5
54
55 56 1.0 57 1.5
56
57 58 1.0
58
C H P N F S O 0
6-31G(d)
****
Cl
6-31+G(d)
****
Ru 0
SDD
****
Ru 0
SDD
Thanks and regards,
Gayani
#8 Re: Multiwfn and wavefunction analysis » Predicting heat of vaporization and heat of sublimation » 2021-04-03 08:30:30
Hi
I have performed a few calculations
1. Overall surface area: 370.72232 Bohr^2 ( 103.81284 Angstrom^2)
2. Overall variance (sigma^2_tot): 0.00078572 a.u.^2 ( 309.39126 (kcal/mol)^2)
3. Product of sigma^2_tot and nu: 0.00010927 a.u.^2 ( 43.02795 (kcal/mol)^2)
As I understood these are the numbers I need to calculated HOS.
Is it correct?
A=103.81284
sigma^2_tot and nu =43.02795
Thanks a lot
#9 Re: Multiwfn and wavefunction analysis » dissociation asymptote » 2021-03-31 05:15:59
Yes I need to calculate BDE
So I will use Sum of electronic and thermal Enthalpies of radicals and molecule and get the difference
Thanks a lot
#10 Quantum Chemistry » convergence problems in Restricted open shell calculation » 2021-03-31 04:51:15
- shenaya
- Replies: 1
Dear All,
I'm performing optimization and frequency calculation on the below system.
It gives a convergence issue.
I have tried several options
ex: increasing the number of cycles.
As we can not use qc, I have used alternative keywords; use=l506
Nothing worked. Your suggestions will be helpful.
Thanks and regards
Shen
opt=calcfc rocam-b3lyp/6-31g(d) scrf=(iefpcm,solvent=water)
scf=maxcycles=125 iop(5/13=1)
Title Card Required
1 2
C 1.35656100 1.13118500 -0.07295400
H 1.28256500 2.07700700 -0.11348300
C 0.90312800 -0.99348200 -0.18842500
H 0.44545800 -1.81385500 -0.32705900
N 0.35868700 0.24094300 -0.38379100
N -4.53096900 0.01670200 0.10290700
H -3.92323500 0.41587700 -0.42267400
H -5.30860900 -0.19732300 -0.32006000
H -4.70348900 0.60048000 0.77614600
H -4.14393200 -0.74142100 0.41322600
N 2.42986200 0.46674300 0.28898400
N 2.14143500 -0.89110000 0.21506800
#11 Multiwfn and wavefunction analysis » Predicting heat of vaporization and heat of sublimation » 2021-03-30 09:14:39
- shenaya
- Replies: 5
Dear All,
I'm calculating the heat of formation of some molecules using CBS-QB3 method. I have few questions.
1. When I'm finding enthalpy of formation of a radical ex: NH2 radical should I use unrestricted CBS-QB3?
2. As described in section 3.15 Quantitative analysis of molecular surface (12) in the manual heat of sublimation will be calculated.
My question is which basis set should use to calculate the Heat of sublimation parameters A and sigma.
CBS-QB3 or B3LYP/6-311++G(2df,2p)
Thanks and regards
Shen
#12 Re: Multiwfn and wavefunction analysis » dissociation asymptote » 2021-03-30 09:03:02
Yes I need to calculate BDE
So I will use Sum of electronic and thermal Enthalpies of radicals and molecule and get the difference
Thanks a lot
#13 Re: Multiwfn and wavefunction analysis » dissociation asymptote » 2021-03-30 01:53:47
Dear Tian,
When using enthalpy shall I use enthalpy of formation or just the enthalpy (Sum of electronic and thermal Enthalpies)
#14 Re: Multiwfn and wavefunction analysis » dissociation asymptote » 2021-03-04 00:46:58
Thank you very much for your comment !!!
#15 Re: Multiwfn and wavefunction analysis » dissociation asymptote » 2021-03-03 07:48:42
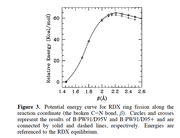
Actually, I'm using this method to calculate bond dissociation energy since my complexes are salts. This salt optimization in the gas phase is very difficult. So I have incorporated the solvent effect.
My question is after dissociation can I check whether it has formed open-shell fragments using multiwfn
#16 Multiwfn and wavefunction analysis » dissociation asymptote » 2021-02-25 16:27:59
- shenaya
- Replies: 8
Dear all,
I'm calculating the bond dissociation energy of a complex system, hence, homolysis bond dissociation can not be used.
I used the following method
ex: if my molecule in CH4 change on of the C-H bond and calculate energy
The plot of Energy vs C-H distance gives BDE at one point.
My question is whether we can find the details of dissociation asymptote to make sure that the correct open-shell fragments have obtained using multiwfn.
Thanks and regards.
#17 Re: Multiwfn and wavefunction analysis » Unit conversion ofelectron density, Laplacian of electron density,V(R) » 2020-12-18 01:35:32


I referred this paper
#18 Re: Multiwfn and wavefunction analysis » Unit conversion ofelectron density, Laplacian of electron density,V(R) » 2020-12-18 00:52:07
Can you please comment on other two units as well
Laplacian of elctron density e nm-5
V(R) eV nm-3
What should be the correct units for these two?
#19 Multiwfn and wavefunction analysis » Unit conversion ofelectron density, Laplacian of electron density,V(R) » 2020-12-17 01:44:18
- shenaya
- Replies: 4
Dear All,
As state in the manual, all the units are in a.u unless specified.
Electron density = 0.328126182
=0.328126182/0.0529^3 e nm-3
Laplacian of electron density =-0.572235415
= -0.572235415/(0.0529^5) e nm-5
V(R) =-4.89E-01
= (-4.89E-01*27.211)/(0.0529^3) eV nm-3
as
1 a.u eV
1 27.211
1 Bohr nm
1 0.052917
Can you please let me know whether the above unit conversion is correct or not?
Can I write electron density as "e nm-3" ???
thanks and regards,
Shen
#20 Re: Multiwfn and wavefunction analysis » Laplacian electron density of energertic materials » 2020-09-24 15:26:47
Thank you very much for your suggestion. I will definitely try this now
#21 Re: Multiwfn and wavefunction analysis » Laplacian electron density of energertic materials » 2020-09-24 05:51:10
In these molecules, N-NO2 is the trigger bond. So I've obtain the bond critical point between N-NO2 and obtained the properties at BCP.
In this article "Charge Density Distribution, Electrostatic Properties, and Impact Sensitivity of the High Energetic Molecule TNB: A Theoretical Charge Density Study.
The most sensitive bond is studied using laplacisan electron density.


As per attached discussion they have identified the weakest bond using laplacian electrons density, Gr, Vr and Hr.
#22 Multiwfn and wavefunction analysis » Laplacian electron density of energertic materials » 2020-09-24 03:19:53
- shenaya
- Replies: 4
Dear All,
I'm performing a study on the impact sensitivity of the energetic materials using AIM analysis. Here I'm using zwitterionic salts and to optimize the molecules I've used IEFPCM=water as the solvent. Gas phase optimizations are not successful as the H atom tend to dissociate and attache to O or N
CAM-B3LYP/6-31g(d)/IEFPCM=water
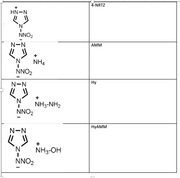
However I can not observe the desired trend from the laplace of electron density analysis.
electrondensity laplace Sensitivity rank
NRTZ 0.394566243 -0.817435389 1
AMM 0.396351132 -0.80044764 3
Hy 0.386374712 -0.76654905 2
HyAm 0.38592834 -0.765011656 2
I expected highest negative value from least sensitive molecule and vise versa.
Hy and HyAm values are ok
NRTZvalue is overestimated.
Can you please comment on this.
Thanks,
Shen
#23 Re: Multiwfn and wavefunction analysis » Critical points with different basis sets » 2020-05-12 00:43:50

Is it possible to generate this type of MO diagram
#24 Re: Multiwfn and wavefunction analysis » Critical points with different basis sets » 2020-05-11 03:47:09
Hi Tian,
I'm performing further analysis of this system. I have calculated MO (HOMO,HOMO-1,LUMO,LUMO+). To discuss the results I need to find which type of orbitals of the LIGAND (sigma, sigma*, pi, pi*) contributed to HOMOs and Lumos. I have already calculated MO % contribution from each atom.
I already went through section 4 and couldn't find it.
Can you please direct me on how to find this information.
Calcuation were done using Gaussian.
Thanks,
Shen
#25 Re: Multiwfn and wavefunction analysis » Critical points with different basis sets » 2020-01-14 02:21:57

Hi Tian,
I have analysed another complex which has Rh(III) which gives more than one bond critical point between Rh and N atoms.
I'm not sure what it really means?
If possible please explain this.
#26 Re: Multiwfn and wavefunction analysis » Critical points with different basis sets » 2020-01-06 08:27:50
Thank you very much for your quick reply.
Problem solved !!!
regards,
Shen
#27 Re: Multiwfn and wavefunction analysis » Critical points with different basis sets » 2020-01-06 01:26:35
Hi Tian,
I've just emailed relevant files to sobereva@sina.com <sobereva@sina.com>;
Please take a look.
best regards,
Shenaya
#28 Re: Multiwfn and wavefunction analysis » Critical points with different basis sets » 2020-01-02 05:57:26

Than you very much for your suggestion
I have performed the calculation using def2-TZVPP basis set.
In the neutral Ru complex no bond path and bond critical point between Ru and F. But in the cationic compound, we can see. The main concern is contour lines are very similar between Ru and F atoms in both of the cases. What are the other parameters that I can use to explain this behaviour?
regards,
Shen
#29 Multiwfn and wavefunction analysis » Critical points with different basis sets » 2019-12-23 02:00:55
- shenaya
- Replies: 12
Dear All,
I have used three different basis sets to obtain critical points. 
These are two Ru(II) complexes. I have obtained critical points and contour plots using above-mentioned basis sets. I'm interested in Ru-F bond
I have a few questions to ask.
1. Although Cationic and Neutral compounds have very similar contour plots,
Neutral Cation
6-311g(d,p) no bond path and bond critical point have a bond path and bond critical point
6-311g(2d,p) no bond path and bond critical point have a bond path and bond critical point
cc-pvtz no bond path and bond critical point no bond path and bond critical point
Is there a threshold value that use multiwin program to define a bond path and critical points?
I'm struggling to explain these results.
Can you refer me further analysis or explain this matter if possible
Thanks and regards,
Shen
#30 Re: Multiwfn and wavefunction analysis » Electron density and Laplacian of electron density » 2019-10-17 01:41:16
Thank you very much for your reply.
Can suggest me some examples or tutorials to follow.
Regards,
Shen