Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Multiwfn and wavefunction analysis » good practice in computing atomic free volume and polarizability » 2023-07-04 11:56:55
- amin.alibakhshi@hotmail.c
- Replies: 0
Dear all,
I recently tried to use Multiwfn to compute C6 parameters based on Tkatchneko-Scheffer method. Nevertheless, for the same dataset, the values I get are found to be by a factor of two less accurate than what they reported in their original paper.
The reason seems to be due to the computation of free atom volumes either via G16 or the default free atom spherical average densities hard-coded in Multiwfn (the same issue also has been discussed for Q-Chem code https://manual.q-chem.com/5.4/sect_TS.html)
For that, I would be so thankful for some hints for good practice in computations leading to more accurate estimation of free-atom volumes for open-shell atoms, specifically about using symmetry or not, computing based on restricted open-shell or unrestricted, and setting initial guesses.
Thank you in advance and best regards,
Amin
#2 Re: Multiwfn and wavefunction analysis » some potential bugs » 2023-06-19 11:34:07
I cannot make it interactively an have to use scripts because I am dealing with thousands of compounds. But I did some quick fix in my scripts via copying H+1.rad file from example/atmrad directory to the local atmrad directory and it fixed the problem and seems like a general remedy because for H+1 it makes no dependence on method/level of theory.
Thanks again for the fantastic job and kind regards,
Amin
#3 Re: Multiwfn and wavefunction analysis » some potential bugs » 2023-06-19 09:55:51
thank you for your quick follow-up. About g16, you were right and it was a bug in my own code. Nevertheless, for H+1, I still have a problem with computations that are automatically done by Multiwfn. I could manually fix it by writing H+1.rad with only one entry "0" and place in atmrad director as you suggested but for automatic usage of Multiwfn, I still get an error for every molecule in which the H atom is present as can be seen in this screenshot:
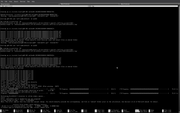
#4 Multiwfn and wavefunction analysis » some potential bugs » 2023-06-18 16:12:57
- amin.alibakhshi@hotmail.c
- Replies: 4
Dear Prof. Lu, Dear developers of Multiwfn,
I just recently noticed two issues that seems to me like a bug.
In the calculation of Fuzzy atomic space analysis based on the Hirshfeld-I method, for cases Gaussian is needed to compute .rad files, while g09 works perfectly, it doesn't work for the g16 version and I suppose the problem stems from the way .gjf is written.
The second issue is related to the same computations when H atom is involved. For that, some errors are generated for H+1 computations which is kind of natural as there is no electron and therefore, wfn or .rad file is not defined in theory.
Kind regards + thank you for your wonderful job
#5 Re: Multiwfn and wavefunction analysis » some potential bug in units? » 2023-06-02 09:28:07
Dear Tian,
many thanks, everything is crystal clear. Just one quick thing: I suppose "dr" in these equations should be dr^3. Am I right?
Best,
Amin
#6 Re: Multiwfn and wavefunction analysis » some potential bug in units? » 2023-05-31 12:26:56
Dear Tian,
many thanks for your response. I am wondering, shouldn't the wavefunctions used to compute free volume and also Hirshfeld weights be (preferably) computed based on the same level of theory to yield consistent results?
The second question is about the equation of q_A in page 78 of manual (first formula in section 3.9.1 Hirshfeld atomic charge). I suppose in that formula the weight only should be multiplied by rho(r) and not rho_diff (i.e. rho^pro should not be weighted). What do you think?
Best regards,
Amin
#7 Re: Multiwfn and wavefunction analysis » some potential bug in units? » 2023-05-25 11:49:24
Dear Tian,
I also have another question. Whenever we try to calculate atomic effective volume, the software in the first step asks for wfn for individual atoms. Then, once all wfns are given, then it searches for wfn of the exact same atoms (for which wfn has already been given bu user) in atomwfn and if it cannot find them, ask for the path of Gaussian. I suppose there should be no difference between the two sets of wfn, but since the software asks for them two times separately, I am wondering, shall they be computed differently?
Best,
Amin
#8 Re: Multiwfn and wavefunction analysis » some potential bug in units? » 2023-05-23 08:30:21
Dear Tian,
many thanks for the information and quick consideration.
Best regards,
Amin
#9 Multiwfn and wavefunction analysis » some potential bug in units? » 2023-05-22 16:58:37
- amin.alibakhshi@hotmail.c
- Replies: 8
Dear All,
for computation of free and effective volume and polarizability of atoms based on Tkatchenko-Scheffler method ( 15-Fuzzy atomic space analysis->option13), the output printed by the software indicates that the computed volumes are in a.u., nevertheless, they seem pretty much to be in Angstrom.^3 unit (for free volumes, computed radii based on spherical shape assumption perfectly matches the vdW radii in Angstrom ). Is it a bug or I got something wrong?
Best,
Amin
#10 Re: Multiwfn and wavefunction analysis » electron density data and isodensity surface position in xyz format » 2023-03-15 10:46:29
Dear Tian,
many thanks for the great help. I though it is just some charge fraction. Everything is clear. Thanks again for the great job,
Best regards,
Amin
#11 Re: Multiwfn and wavefunction analysis » electron density data and isodensity surface position in xyz format » 2023-03-14 14:59:20
Dear Tian,
many thanks for your response and also for your fantastic software, really appreciate it.
I just tried main function 5 and computed electron densities to the medium quality grid and exported it to output.txt. Nevertheless, the sum of densities (values in the fourth column) seems to be quite large (ca 6700) while the total number of electrons in my system is 152. Am I missing something or should I normalize it on my own?
Best,
Amin
#12 Multiwfn and wavefunction analysis » electron density data and isodensity surface position in xyz format » 2023-03-14 14:14:24
- amin.alibakhshi@hotmail.c
- Replies: 4
Hi All,
I need electron density data in xyz format. Multiwfn computes and use it frequently (e.g. in Quantitative analysis of molecular surface) but I couldn't find a way to obtain and save such data (the electron density in cartesian coordinates around the molecule preferably for some given data points as well as the cartesian coordinate of isodensity surfaces). I would be so thankful for any hint.
Best regards,
Amin
#13 Re: Multiwfn and wavefunction analysis » computation of atomic radii and molecular surfaces via QTAIM » 2022-09-06 14:51:34
I found what I was looking for and noticed it is already implemented in the Multiwfn (e.g. example of page 699).
Many thanks for developing such an amazing code
Best,
Amin
#14 Re: Multiwfn and wavefunction analysis » computation of atomic radii and molecular surfaces via QTAIM » 2022-09-06 13:46:38
Dear Amin,
It is possible to calculate radius of isolated atom/ion using Multiwfn based on isosurface of rho=0.001 a.u., however this is not directly applicable to atoms in molecular environment.
Best,
Tian
Dear Tian,
Can some sort of on-the-fly optimization of atomic radii in a way to give the highest overlap between the generated vdW surfaces using those radii and calculated basins, be a potential solution?
I would highly appreciate your comments on that.
Best,
Amin
#15 Re: Multiwfn and wavefunction analysis » computation of atomic radii and molecular surfaces via QTAIM » 2022-09-06 13:36:10
Dear Amin Alibakhshi,
I don't well understand your problem. AIM doesn't provide an unambiguous definition of atomic radius and surface.
Best,
Tian
thanks a lot prof. Lu for your response. I had read some papers which used AIM to compute atomic radii but I cannot find them. Apart from using AIM, can Multiwfn somehow give a quantum mechanically computed estimation of atomic radii? I heard it is possible but I cannot find much details on it in the manual.
Best,
Amin
#16 Multiwfn and wavefunction analysis » computation of atomic radii and molecular surfaces via QTAIM » 2022-09-06 09:01:22
- amin.alibakhshi@hotmail.c
- Replies: 5
Hi everyone,
I have a question regarding the quantum theory of atomes-in-molecules implemented in Multiwfn.
I am interested to approximate the total surface of a molecule as well as an estimation of atomic radii in a specific molecule based on this method/basin analysis. I would be so thankful for any hint.
Best regards,
Amin Alibakhshi
#17 Re: Multiwfn and wavefunction analysis » Evaluate contribution of energy from a single molecule » 2020-11-30 16:50:02
thanks so much
#18 Re: Multiwfn and wavefunction analysis » Evaluate contribution of energy from a single molecule » 2020-11-21 11:21:58
Hello,
In principle it is possible, you simply need to sum up atomic contributions as molecule contribution.
many thanks prof. Lu both for your answer and for your super great job
one last question, is it already implemented and can be carried in Multiwfn? If yes, I would appreciate some few hints
Best regards,
Amin
#19 Multiwfn and wavefunction analysis » Evaluate contribution of energy from a single molecule » 2020-11-20 13:10:13
- amin.alibakhshi@hotmail.c
- Replies: 5
Hi everyone,
I am wondering, is there any possibility to evaluate/approximate the total energy of a single molecule in a cluster of few molecules via Multiwfn (contribution of a single molecule into the total energy of the cluster which we calculate via QM) ? (something like calculating per atom energies via energy decomposition analysis but instead of atom, this time for a whole molecule).
thanks in advance for any help.
Best regards,
A.
#20 Re: Multiwfn and wavefunction analysis » error forrtl: severe (174): SIGSEGV, segmentation fault » 2019-08-25 15:13:19
Many thanks Tian Liu, for your prompt help and also the wonderful work.
the "ulimit -s unlimited" solved the problem, as you said :-)
thanks again.
#21 Multiwfn and wavefunction analysis » error forrtl: severe (174): SIGSEGV, segmentation fault » 2019-08-25 12:46:53
- amin.alibakhshi@hotmail.c
- Replies: 4
Hi all,
I am new in Multiwfn and am trying to run the first job which is a RESP point charge calculation. As soon as I type Multiwfn and run the code in Linux, for the next step I type the fchk name "1.fchk" and after few second the error "error forrtl: severe (174): SIGSEGV, segmentation fault" and the Multiwfn stops.
I am using the latest version of the Multiwfn on a Lunix HPC cluster.
Do you have any idea how to fix this issue?
Thanks in advance for your help.
Amin
Pages: 1