Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » Calculation of Pauli potential using multiwfn » 2025-08-28 04:48:19
Thank you very much for the detailed explanation and for pointing me to the relevant section of the manual.
#2 Re: Multiwfn and wavefunction analysis » Calculation of Pauli potential using multiwfn » 2025-08-27 12:44:44
Thank you very much for your reply and for sharing the details. I have followed the same method as you suggested and obtained almost the same graph. However, I observe negative values on the order of 10⁻⁴ near zero, along with some irregular behavior in that region.
If I want to change the number of grid points and the length of the radius, what is the correct way to do so? I suspect that increasing the number of grid points might help resolve this abnormality near zero.
Also, I apologize for these are very basic questions — I am quite new to using Multiwfn.
#3 Re: Multiwfn and wavefunction analysis » Calculation of Pauli potential using multiwfn » 2025-08-27 09:34:01

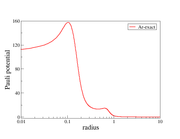
This is the Pauli potential graph I obtained by following the steps discussed above in Multiwfn. The other image I am attaching here corresponds to the Pauli potential obtained from my manual calculation. I would kindly request if you could try running the Pauli potential calculation for the Ar atom using the same procedure and share the results with me, so that I can cross-check my implementation.
#4 Re: Multiwfn and wavefunction analysis » Calculation of Pauli potential using multiwfn » 2025-08-27 06:59:07
Thank you for the suggestion. However, my intention is not to check whether the Pauli potential is spherically symmetric or not. What I actually want is to calculate the Pauli potential of atoms and molecules and save its values along the radial direction.
#5 Re: Multiwfn and wavefunction analysis » Calculation of Pauli potential using multiwfn » 2025-08-25 09:47:03
%mem=90mw
%nprocs=23
%chk=Ar.chk
# HFS/6-31G output=wfn Pop =full
gfinput gfoldprint iop(6/7=3)
title card
0 1
Ar 0.000000 0.000000 0.000000
Ar.wfn
This is the Gaussian input file I used to run my calculation. After that, I loaded the Ar.fchk file into Multiwfn. From the main menu, I selected option 3 (“Output and plot specific property in a line”). Then I chose option 100 (“User-defined function (iuserfunc = 60), see Section 2.7 of the manual”).
After this, the following prompt appeared:
0 Set extension distance for mode 1, current: 1.5000 Bohr
1 Input index of two atoms to define a line
2 Input coordinate of two points to define a line
At this stage, I selected option 2 and entered the x, y, and z coordinates as 0,0,0, 10,0,0.
#6 Re: Multiwfn and wavefunction analysis » Calculation of Pauli potential using multiwfn » 2025-08-21 05:18:58
I have used this information given below on page 39 multiwfn manual Multiwfn_3.8_dev. 60 Pauli potential: V. Multiwfn supports two ways to evaluate this function, depending on
"ispecial" in settings.ini:
• ispecial=0: According to Eq. 17 of Comput. Theor. Chem., 1006, 92 (2013), V = + VESP − VXC
− VW, where is chemical potential (assumed to be zero by Multiwfn), VESP is electrostatic potential
(as described in Section 2.6) and VXC is exchange-correlation potential (its form is determined by
"iDFTxcsel" in settings.ini, see later).
• ispecial=1: Use original definition, namely ?θ = ?S − ?W = ??S
[?]/?? − ??W[?]/??, where VS is potential of non-interacting kinetic energy functional (S), and VW is potential of Weizsäcker
kinetic energy functional (vW). The form of S can be chosen by "iKEDsel" in settings.ini, see later;
currently only iKEDsel of 3, 5, 7 are supported
#7 Multiwfn and wavefunction analysis » Calculation of Pauli potential using multiwfn » 2025-08-20 17:49:48
- Aparna Gangwar
- Replies: 12
I have manually calculated the Pauli potential and electron density for atoms with spherically symmetric ground-state densities using data from Gaussian log files. To verify my results, I also attempted to calculate the Pauli potential using Multiwfn. I carefully followed the steps provided in the Multiwfn manual, but the values I obtained from the software do not match my manually calculated results.Could anyone please clarify the correct procedure for calculating the Pauli potential in Multiwfn?
Pages: 1