Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » Aromaticity index calculation » 2026-05-13 13:56:41
Dear Prof Tian Lu,
Simply Amazing!! thank you so much for your detailed response !!
My best
Alessio
#2 Multiwfn and wavefunction analysis » Aromaticity index calculation » 2026-05-12 20:09:23
- alessiomacorano
- Replies: 2
Dear Prof. Tian Lu,
First of all, thank you very much for your outstanding work on MULTIWFN and for managing the forum.
I have two quick questions:
1) In your opinion, what are the most appropriate aromaticity indices for studying π-interactions in benzene dimers?
(The example geometries are the three shown in the image)
2) If I wanted to study the aromaticity of a single benzene ring, which indices would you recommend?
I ask because HOMA and the Bird index come to mind, but perhaps they aren’t sufficient.
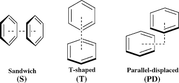
Thank you so much for your Help!
My best
Alessio
#3 Re: Quantum Chemistry » Frequency calculation in benzene dimer π–π stacking » 2026-04-14 06:46:30
Dear Prof Tian Lu,
Thank you so much, this clears up the questions I had.
Thank you very much.
Alessio Macorano
#4 Re: Quantum Chemistry » Frequency calculation in benzene dimer π–π stacking » 2026-04-13 12:41:23
Dear Prof Tian Lu,
So if I understand correctly, I have to perform opt+freq for the structure corresponding to the step 1 as reported in the image ?
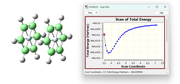
Finally, as a more general question, in this seminal work: https://doi.org/ 10. 1021/ ja025896h, as well as in other works by Sherrill David, it appears that the binding energy was calculated solely from the optimization of the inter-monomeric distance; I, too, optimized only the distance between the monomers in my calculations. I would kindly ask for your opinion on this, because I am a bit confused as to whether, in the calculations of binding energy for π-π interactions, the frequencies should be calculated or not at this point.
Thank you so much
my best
Alessio
#5 Quantum Chemistry » Frequency calculation in benzene dimer π–π stacking » 2026-04-10 09:49:22
- alessiomacorano
- Replies: 4
Dear Prof. Tian Lu,
First of all, thank you very much for your outstanding work on MULTIWFN and for managing the forum.
I am currently working on modeling π-π interactions between benzene dimers (Gaussian 16), for example in the “face-to-face” (FtF) geometry as shown in the attached figure. I performed the relaxed scan by keeping the monomer geometry fixed and optimizing the distance between them. I think I made a mistake because in the route section I used:
opt=modredundant gen nosymm geom=connectivity counterpoise=2 blyp and mpiricaldispersion=gd3 int=grid=superfine
without including the frequency calculation. So, to “fix” the error, I was thinking of saving the minimum geometry obtained from the relaxed scan and calculating the vibrational frequencies based on that with the freq keyword. Is this the correct procedure in your opinion?
If I obtain negative vibrational frequencies, how should I proceed?
Thank you in advance,
Alessio
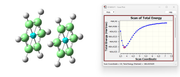
#6 Re: Multiwfn and wavefunction analysis » SAPT analysis in MULTIWFN » 2026-02-19 08:21:41
Dear Tian Lu
This is great, I will check on it.
Thank you so much
my best,
Alessio
#7 Multiwfn and wavefunction analysis » SAPT analysis in MULTIWFN » 2026-02-17 15:35:36
- alessiomacorano
- Replies: 2
Dear Prof Tian Lu
First of all thank you so much for all your work of Multiwfn.
Actually, I'm working on modelling the π–π stacking between benzene dimers using the DFT (level of theory: 6-311+g(2d,2p) blyp empiricaldispersion=gd3), I would like to simply ask if there is any way in MULTIWFN to perfrom SAPT analysis on the optimized geometry?
I heard about the EDA energy decomposition analysis but I don't know the differences with SAPT.
Do you might have any suggestion ?
Thank you so much,
My best
Alessio
#8 Re: Multiwfn and wavefunction analysis » Nucleophilic idenx calculation in Multiwfn » 2025-09-10 14:44:45
Dear Prof. Tian Lu,
Thank you very much for your clear answer. So, for a set of different molecules that have the same nucleophilic reactive group, for example -SH (as in my case), can I also use Hirshfeld atomic charges or the Fukui function to evaluate the nucleophilicity of sulfur S between different molecules and make a rank between the molecules from the most nucleophile to the less one right ?
my best
Alessio
#9 Multiwfn and wavefunction analysis » Nucleophilic idenx calculation in Multiwfn » 2025-09-04 09:23:17
- alessiomacorano
- Replies: 3
Dear Prof Tian Lu
First of all thank you so much for all your work of Multiwfn.
Actually, in collaboration with an experimental lab we are working on the dissociation reaction (retro michael reactions) of some thiols and some unsaturated aldehyde compounds such as crotonaldehyde. The experimental conditions are set at a temperature of 37° Celsius at pH 7.4 to study the dissociation reactions. I'm performing DFT calculations to support experiments.
I have a set of some thiolates molecule that I have optimized using the folowing level of theory: 'opt=calcfc freq=noraman wb97xd/6-311+g(d,p) scrf=(cpcm,solvent=water)', I have done the same also for TCE molecule as reference for calculate the nucleophilicity index.
I have used the implicit solvent and I have carried out the calculation of the nucleophilic index on the atttached thiolate (cysteamine) in the protonated form since the experiemental condition is pH 7.4 becuase we wanted to correlated kinetic constant as kon with nucleophilicity index so,
If possible, I have a couple of questions:
1) Since I wanted to correlate with experimental data is right to include the implicit solvent in the calculation for the Nucleophilic index ? or just considering the gas phase ?
2) Is it correct to calculate the nucleophilicity index considering the experimental conditions (for example considering the protonated amine NH3 since it's the same compund in the experiemntal lab), given that I want to compare it with experimental data? Is the nucleophilic index accurate for charged molecules ? or should I use another index?
Thank you so much,
My best
Alessio
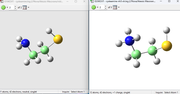
#10 Re: Multiwfn and wavefunction analysis » NBO population analysis bond order MULTIWFN » 2025-06-10 07:53:12
Dear Prof Tian Lu,
Thank you so much for your reply, it helps me a lot !
My best
Alessio
#11 Multiwfn and wavefunction analysis » NBO population analysis bond order MULTIWFN » 2025-06-09 14:43:38
- alessiomacorano
- Replies: 2
Dear Prof Tian Lu,
First of all, thank you very much for your outstanding work on the Multiwfn software.
I have a question regarding bond order analysis. In the paper "Regio-selectivity prediction with a machine-learned reaction representation and on-the-fly quantum mechanical descriptors", the authors used the keyword pop=(full,mbs,hirshfeld,nbo6read) in Gaussian to extract NBO-based bond orders via the licensed NBO 6.0 program.
Is it possible to obtain similar bond order information from NBO population analysis using Multiwfn instead?
My best
Alessio
#12 Re: Multiwfn and wavefunction analysis » Population analysis » 2024-05-10 12:47:31
Dear Prof Tian Lu,
Thank you so much for your reply that's clarified a lot for me !!
Thanks a lot
Have a good week-end
My best
Alessio
#13 Multiwfn and wavefunction analysis » Population analysis » 2024-05-10 10:01:55
- alessiomacorano
- Replies: 3
Dear Prof Tian Lu,
I'm wondering if I could ask you one question about the population analysis, because i'd like to obtain the partial charges.
Is the QTAIM very reliable respect to the NPA, Mulliken and NBO analysis ?
moreover considering one molecules and trying different population analysis methods, how to know whcih could be the best ?
Thank a lot
My best
Alessio Macorano
#14 Re: Multiwfn and wavefunction analysis » molecular descriptors for electron densities » 2024-01-31 01:48:28
Dear Professor Tian Lu,
Thank you so much
I will check in that section.
Best
Alessio
#15 Multiwfn and wavefunction analysis » molecular descriptors for electron densities » 2024-01-18 17:43:30
- alessiomacorano
- Replies: 2
Dear Prof Tian Lu
Regarding to Electron density we are used to visualize them for a molecule, I would like to ask you how can I instead obtain a molecular descriptors to quantify the electron density, clearly I think that the total electron density cannot be quantified, but maybe in specific regions like HOMO and LUMO, yes.
Probably I miss to see the example in MULTIWFN.
Thank you for your work !!
Alessio
#16 Re: Multiwfn and wavefunction analysis » Polar surface area » 2024-01-12 16:48:49
Dear Prof Tian Lu, probably I think that the different value from Multiwfn and pubchem are different because from pubchem the polar surface area is calculated with Cactvus software (which probably, I'm checking in the meantime) used the topological polar surface area defined by Peter ERTL in his paper.
Probably this can take into account for the difference.
Regarding to the Polar surface area from MULTIWFN do you have more reference apart the manual, I ask you for the ongoing publication.
Thank you so much !!
Alessio Macorano
#17 Multiwfn and wavefunction analysis » Polar surface area » 2023-12-19 21:24:39
- alessiomacorano
- Replies: 3
Dear Prof Tian Lu
I would like to ask you one question about the topological surface analysis from .fchk of Gaussian or related QM software, the .fchk are generated from chk with b3lyp/6-311++g(d,p) with SMD implicit model solvent water.
On some drug like molecules i would like to use PSA (polar surface area) as molecular descriptors.
To do this i tried to calculate with multiwfn software from .fchk of Gaussian 16 software, but I obtain different values of PSA respect to Pubchem for example, and I don't understand why.
Maybe the difference is for the implicit model solvent ? should i have to consider in gas phase ?
Thank you so much for your work and this forum
Alessio
Pages: 1