Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » Simulation of pre-resonance Raman spectrum » 2024-10-16 14:20:20
Dear Tian,
Many thanks for the reply. I will try with the Gaussian!
#2 Multiwfn and wavefunction analysis » Simulation of pre-resonance Raman spectrum » 2024-10-15 19:20:54
- zmfhtm2045
- Replies: 2
For the mistake about previous question, I editted the question.
Hi, Tian. First, thank you for sharing this great software.
I am now trying to simulate pre-resonance raman spectrum from the Orca.
I made input following 3.13.2 in manual using numfreq keyword. And I get a .out file from the calculation results.
But, when I open .out file through Multiwfn, there are no options for setting wavelength of incident beam.
Qustion. How to set the any incident light wavelength by myself to perform pre-resonance Raman simulations in Multiwfn?
Here, I copied my input file for the calculation of orca.
The Cartesian Coordinate of this file came from Opt Freq calculation. I want to set incident beam wavelength as 460 nm.
! numfreq PBE0 def2-TZVP def2/J RIJCOSX tightSCF CPCM(water)
%maxcore 6300
%PAL NPROCS 28 END
%elprop Polar 1 end
* xyz 0 1
#3 Quantum Chemistry » UV/Vis absorption simulation of diradicals » 2024-09-11 18:27:08
- zmfhtm2045
- Replies: 2
Hello, I am studying for the organometallic cobalt complex having diradical characteristics (experimentally observed).
The thing I want to do is simulation of UV-Vis absorption spectra simulation about singlet diradical organometallic compound.
Could I simulate that by using Orca and Multiwfn?
If possible, how can I set the keyword for the Orca? I tried spin-flip TDDFT calculation for the compounds but, it fails with a error message (Error: CSI/TDDFT) ... aborted.)
Here, my last trial of input files.
!B3LYP DEF2-SVP CPCM(acetonitrile) PAL8
%maxcore 8000
%TDDFT
SF TRUE
NROOTS 30
END
* xyz 3 1 Co.xyz *
#4 Re: Multiwfn and wavefunction analysis » Natural Transition Orbital (NTO) overlap density plots » 2023-04-17 11:33:01
Dear friends,
I found this method in 3.16 section in the manual. Thanks.
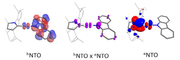
#5 Multiwfn and wavefunction analysis » Natural Transition Orbital (NTO) overlap density plots » 2023-04-17 07:37:51
- zmfhtm2045
- Replies: 1
Hello everyone,
I am a newbie in using Multiwfn. Now, I have conducted for TD-DFT calculations with Gaussian, and I am currently working on calculating NTOs for small molecules with donor-acceptor characteristics.
I would like to generate isosurface plots showing the product between NTO(hole) and NTO(electron) as described in the paper (J. Am. Chem. Soc. 2019, 141, 8616-8626) (refer to the attached picture for the center of NTO overlap).
My questions are:
Does Multiwfn support functions for plotting newly defined grids?
If so, how can I generate the density plot for the product between NTO(hole) and NTO(electron))?
Thank you!
Pages: 1