Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
- Index
- » Search
- » Posts by Dirk
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » gradient of ESP » 2025-07-29 04:07:27
Hello again,
I am plotting 2D ESP maps of a molecule, derived from a wfn file.
The succession in menu numbers is 4 - 12 - 6 which creates an ESP map overlaid by gradient lines that can display
directional arrowheads on the gradient line (which currently point to the opposite direction of the e-field vectors).
In the additional setting I have enabled filling the area between the ESP contour lines with colors and the color index is
then mapped to a scale bar on the right side of the plot. Please have a look at the image of a LiF molecule.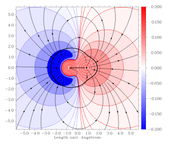
#2 Re: Multiwfn and wavefunction analysis » gradient of ESP » 2025-07-28 15:38:04
This does not help because the scale bar for ESP values would still show the wrong (=inverted) values.
I think for the vector field calculation you are offering the possibility to change the direction of the vectors
to align with the e-field. So the functionality is already present and probably only needs to be enabled for
the direction of the gradient lines ?
#3 Re: Multiwfn and wavefunction analysis » gradient of ESP » 2025-07-28 13:59:36
Dear Tian,
thank you for the fast reply. Well, I tried your suggestion, but the multilication also reverses the values for the ESP and accordingly
produces the wrong charge distribution on the molecule. I think that the option has to work on the gadient calculation only.
The electric field (E) is the *negative* gradient of the electric potential (V): E = -∇V.
Best wishes, Dirk
#4 Multiwfn and wavefunction analysis » gradient of ESP » 2025-07-27 09:49:55
- Dirk
- Replies: 7
Hello Tian,
when analyzing *.wfn files and plotting ESP maps there is an option for plotting also the gradient lines for the ESP.
Unfortunately, the direction of the tips on the gradient lines cannot be changed (=reversed).
At present they point from negative to positive values, whereas e-field lines should point to the opposite direction.
Could you please add an option for changing the direction of the tips ?
Thank you !
Best wishes, Dirk
#5 Multiwfn and wavefunction analysis » output of partial charges into pdb or mol2 file » 2021-11-21 15:04:55
- Dirk
- Replies: 1
Hello,
Multwfn offers a host of methods for calculating partial atomic charges. However, is it also possible to
export the generated charges in a commonly used file format such as pdb or mol2 ?
(PS: I am aware of the function to generate a *.chg file which contains a list of xyz coordinates and charges.)
Thanks for support in advance !
Sincerely, Dirk
#6 Multiwfn and wavefunction analysis » DDEC-6 charges » 2021-04-03 08:11:43
- Dirk
- Replies: 1
Dear Tian,
within the many approaches for calculating partial charges, for 3D systems the so calles DDEC charge decomposition
analysis is becoming more and more popular. https://sourceforge.net/projects/ddec/
Is this method under consideration for Multiwfn, too ?
With kind regards, Dirk
#7 Re: Multiwfn and wavefunction analysis » CM 5 charges from xtb » 2021-04-03 07:52:34
Dear Tian,
thank you very much for the fast reply. I am going to post a feature request to the dvelopers of CP2k.
Reading the manual of the recent version of the stand-alone version of xtb it appeared that the GFn-1 version
of the code produces Mulliken & CM-5 charges by default. The GFn-1 functional is capable of treating 3D periodic cell,
hence there exists an acceptable solution.
#8 Multiwfn and wavefunction analysis » CM 5 charges from xtb » 2021-04-02 09:50:52
- Dirk
- Replies: 3
Dear Dr. Lu,
thank you very much for extending population analyses toward periodic systems.
I have a question relating to the calculation of CM5 charges from CP2K files:
CP2k integrates the Gfn-1-xtb tight binding approach from Grimme et al. and
calculation of periodic cells with 100s of atoms are quite fast and efficient.
Could the output from xtb calculations also be utilized to create CM5 charges,
and if so: are large deviations of the values in comparison to DFT calculations to be expected ?
With kind regards, Dirk
Pages: 1
- Index
- » Search
- » Posts by Dirk