Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » Vector field map of ESP - problem with colors » 2022-01-05 13:38:02
I'll try to explain it better, then.
I did a scan of a molecule of HOCl with a variable electric field and I tried to do the vector field of the molecules. Here'sn input example:
%chk=field_1.chk
%mem=8GB
%nprocs=4
#p b3lyp/6-31+G(d,p) Field=z-200 output=wfn EmpiricalDispersion=GD3BJ
Title Card Required
0 1
O 0.03617200 1.10337600 0.00000000
H -0.90429300 1.32914000 0.00000000
Cl 0.03617200 -0.59742000 0.00000000
field_1.wfn
However, all I saw is this 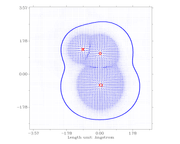 .
.
So, I don't know if there's something I'm doing wrong or not, since the manual does not show something like that.
#2 Multiwfn and wavefunction analysis » Vector field map of ESP - problem with colors » 2022-01-04 22:04:25
- fjbenitez
- Replies: 4
Dear sir:
I applied an electric field on a HOCl molecule along with the reaction coordinate so we could visualize any effect on the sigma hole of Cl. However, when I put them on Multiwfn, I see blue arrows without contrast. Is there something wrong with my version, or should I check something else?
I really appreciate any help you can provide.
#3 Multiwfn and wavefunction analysis » Electron Density Topology Analysis » 2021-06-19 23:31:09
- fjbenitez
- Replies: 1
Dear Sir:
I'm making several electron spin densities of several semiquinones and I wanted to do the topology analysis that appears in the Multiwfn 3.7 manual, but when it comes to the topology analysis itself, it does not show me the same steps. Specifically, my problems start after I export the attractors.pdb file.
Could you please help me?
#4 Re: Multiwfn and wavefunction analysis » Fukui functions » 2021-06-02 20:16:00
Thank you!
And yes, I already did the procedure shown on Section 4.100.16.2 of Multiwfn manual, but I need to compare both methods.
#5 Re: Multiwfn and wavefunction analysis » Fukui functions » 2021-06-01 21:43:33
Dear sir:
Is it possible to calculate Fukui functions and Dual descriptor using HOMO and LUMO densities on Multiwfn?
Thanks in advance!
#6 Re: Multiwfn and wavefunction analysis » Reduce cost of ESP analysis on molecular surface for ORCA users » 2021-01-07 21:48:38
Hi!
I tried to use your suggestion and the program says the following: "Note: Albeit "orca_2mklpath" parameter in settings.ini has been defined, the orca_2mkl executable file cannot be located, therefore the .gbw file cannot be directly opened by Multiwfn".
So, what do I do?
PS: I'm new at this, just trying it for the first time ![]()
Pages: 1