Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Quantum Chemistry » How can i get wfx file for MP2? » 2025-12-18 18:00:00
Thank you very much for your time and support.
#2 Quantum Chemistry » How can i get wfx file for MP2? » 2025-12-18 03:51:56
- choconostle123
- Replies: 2
Hello I hope you are doing well.
I would like to ask about the MP2 WFX file, because I want to use the wavefunction from an MP2 calculation in Gaussian (with the keyword density=current) in AIMAll. However, I am not sure whether I need to modify the file or do anything else. Finally, is it possible to do this using Multiwfn?
Thank you very much in advance for your time and support.
#3 Multiwfn and wavefunction analysis » from fchk produce wfx with virtual orbitals » 2025-04-17 15:05:23
- choconostle123
- Replies: 1
Hello, I want to convert an FCHK file from a Gaussian calculation to a WFX file, including both occupied and virtual orbitals. My question is: is this possible?
Thank you very much for your support Best regards,
#4 Quantum Chemistry » Obtain values of two electron integrals with gaussian09? » 2025-03-26 17:57:41
- choconostle123
- Replies: 1
Hello i want to ask if is there some keyword or method to obtain two electron integrals (ij|kl) from gaussian 09?
#5 Quantum Chemistry » question about the output of state average of casscf on gaussian » 2025-02-07 18:10:14
- choconostle123
- Replies: 1
Hello, I am learning about how to calculate excited states using CASSCF in Gaussian, but I have some doubts about the excitation energy in the output.
I performed a calculation for the formaldehyde molecule with stateaverage . The input is as follows:
#p cas(4,3,NRoot=3,StateAverage) def2tzvp
Title Card Required
0 1
C -0.00001600 0.52685200 0.00000000
H 0.92907900 1.09677600 0.00000000
H -0.92885800 1.09725000 0.00000000
O -0.00001600 -0.66939200 0.00000000
0.3333 0.3333 0.3333
However, in the output, I don't know where the excitation energy is :S.
Are the eigenvalues the energies of the ground state and the 2 excited states?
I am sharing the output below.
-----------------------------------------
#p cas(4,3,NRoot=3,StateAverage) def2tzvp
-----------------------------------------
1/38=1/1;
2/12=2,17=6,18=5,40=1/2;
3/5=44,7=101,16=1,25=1,32=1,116=101/1,2,3;
4/17=4,18=3/1,5;
5/5=2,17=11000000,28=3,38=5/10;
6/7=2,8=2,9=2,10=2,28=1/1;
99/5=1,9=1/99;
Leave Link 1 at Fri Feb 07 11:51:36 2025, MaxMem= 0 cpu: 0.0
(Enter C:\G09W\l101.exe)
-------------------
Title Card Required
-------------------
Symbolic Z-matrix:
Charge = 0 Multiplicity = 1
C -0.00002 0.52685 0.
H 0.92908 1.09678 0.
H -0.92886 1.09725 0.
O -0.00002 -0.66939 0.
NAtoms= 4 NQM= 4 NQMF= 0 NMMI= 0 NMMIF= 0
NMic= 0 NMicF= 0.
Isotopes and Nuclear Properties:
(Nuclear quadrupole moments (NQMom) in fm**2, nuclear magnetic moments (NMagM)
in nuclear magnetons)
Atom 1 2 3 4
IAtWgt= 12 1 1 16
AtmWgt= 12.0000000 1.0078250 1.0078250 15.9949146
NucSpn= 0 1 1 0
AtZEff= 0.0000000 0.0000000 0.0000000 0.0000000
NQMom= 0.0000000 0.0000000 0.0000000 0.0000000
NMagM= 0.0000000 2.7928460 2.7928460 0.0000000
AtZNuc= 6.0000000 1.0000000 1.0000000 8.0000000
Leave Link 101 at Fri Feb 07 11:51:36 2025, MaxMem= 33554432 cpu: 0.0
(Enter C:\G09W\l202.exe)
Input orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 -0.000016 0.526852 0.000000
2 1 0 0.929079 1.096776 0.000000
3 1 0 -0.928858 1.097250 0.000000
4 8 0 -0.000016 -0.669392 0.000000
---------------------------------------------------------------------
Distance matrix (angstroms):
1 2 3 4
1 C 0.000000
2 H 1.089968 0.000000
3 H 1.090001 1.857937 0.000000
4 O 1.196244 1.995637 1.995939 0.000000
Stoichiometry CH2O
Framework group CS[SG(CH2O)]
Deg. of freedom 5
Full point group CS NOp 2
Largest Abelian subgroup CS NOp 2
Largest concise Abelian subgroup C1 NOp 1
Standard orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 -0.000016 -0.526852 0.000000
2 1 0 0.929079 -1.096776 0.000000
3 1 0 -0.928858 -1.097250 0.000000
4 8 0 -0.000016 0.669392 0.000000
---------------------------------------------------------------------
Rotational constants (GHZ): 290.5360811 39.5870535 34.8399315
Leave Link 202 at Fri Feb 07 11:51:36 2025, MaxMem= 33554432 cpu: 0.0
(Enter C:\G09W\l301.exe)
Standard basis: def2TZVP (5D, 7F)
Ernie: Thresh= 0.10000D-02 Tol= 0.10000D-05 Strict=F.
There are 60 symmetry adapted cartesian basis functions of A' symmetry.
There are 24 symmetry adapted cartesian basis functions of A" symmetry.
There are 52 symmetry adapted basis functions of A' symmetry.
There are 22 symmetry adapted basis functions of A" symmetry.
74 basis functions, 118 primitive gaussians, 84 cartesian basis functions
8 alpha electrons 8 beta electrons
nuclear repulsion energy 31.5866087459 Hartrees.
IExCor= 0 DFT=F Ex=HF Corr=None ExCW=0 ScaHFX= 1.000000
ScaDFX= 1.000000 1.000000 1.000000 1.000000 ScalE2= 1.000000 1.000000
IRadAn= 0 IRanWt= -1 IRanGd= 0 ICorTp=0 IEmpDi= 4
NAtoms= 4 NActive= 4 NUniq= 4 SFac= 1.00D+00 NAtFMM= 60 NAOKFM=F Big=F
Integral buffers will be 262144 words long.
Regular integral format.
Two-electron integral symmetry is turned off.
Leave Link 301 at Fri Feb 07 11:51:36 2025, MaxMem= 33554432 cpu: 0.0
(Enter C:\G09W\l302.exe)
NPDir=0 NMtPBC= 1 NCelOv= 1 NCel= 1 NClECP= 1 NCelD= 1
NCelK= 1 NCelE2= 1 NClLst= 1 CellRange= 0.0.
One-electron integrals computed using PRISM.
1 Symmetry operations used in ECPInt.
ECPInt: NShTT= 465 NPrTT= 1428 LenC2= 466 LenP2D= 1323.
LDataN: DoStor=T MaxTD1= 6 Len= 172
The smallest eigenvalue of the overlap matrix is 1.414D-03
NBasis= 74 RedAO= F EigKep= 0.00D+00 NBF= 52 22
NBsUse= 74 1.00D-04 EigRej= 0.00D+00 NBFU= 52 22
Leave Link 302 at Fri Feb 07 11:51:36 2025, MaxMem= 33554432 cpu: 0.0
(Enter C:\G09W\l303.exe)
DipDrv: MaxL=1.
Leave Link 303 at Fri Feb 07 11:51:36 2025, MaxMem= 33554432 cpu: 0.0
(Enter C:\G09W\l401.exe)
ExpMin= 9.52D-02 ExpMax= 2.70D+04 ExpMxC= 9.22D+02 IAcc=2 IRadAn= 4 AccDes= 0.00D+00
Harris functional with IExCor= 205 and IRadAn= 4 diagonalized for initial guess.
HarFok: IExCor= 205 AccDes= 0.00D+00 IRadAn= 4 IDoV= 1 UseB2=F ITyADJ=14
ICtDFT= 3500011 ScaDFX= 1.000000 1.000000 1.000000 1.000000
FoFCou: FMM=F IPFlag= 0 FMFlag= 100000 FMFlg1= 0
NFxFlg= 0 DoJE=T BraDBF=F KetDBF=T FulRan=T
wScrn= 0.000000 ICntrl= 500 IOpCl= 0 I1Cent= 200000004 NGrid= 0
NMat0= 1 NMatS0= 1 NMatT0= 0 NMatD0= 1 NMtDS0= 0 NMtDT0= 0
Petite list used in FoFCou.
Harris En= -113.998494491141
JPrj=0 DoOrth=F DoCkMO=F.
Initial guess orbital symmetries:
Occupied (A') (A') (A') (A') (A') (A') (A") (A')
Virtual (A") (A') (A') (A') (A") (A') (A') (A') (A') (A')
(A') (A") (A") (A') (A') (A") (A') (A') (A') (A')
(A") (A") (A') (A') (A') (A') (A") (A") (A') (A")
(A') (A') (A') (A') (A') (A") (A') (A') (A") (A")
(A') (A') (A") (A') (A") (A') (A") (A') (A') (A')
(A') (A') (A") (A') (A") (A') (A") (A') (A") (A')
(A') (A") (A') (A') (A') (A')
The electronic state of the initial guess is 1-A'.
Leave Link 401 at Fri Feb 07 11:51:36 2025, MaxMem= 33554432 cpu: 0.0
(Enter C:\G09W\l405.exe)
Truncation Level= 99999
a= 2 b= 0 c= 1
a=N/2 - s b=2s c=n- (a+b)
no. active orbitals (n) 3
no. active ELECTRONS (N)= 4
IRREPS TO BE RETAINED = 1 2
GROUP IRREP. MULT. TABLE
1 2
2 1
IRREP. LABELS FOR ORBITALS
2 1 2
BOTTOM WEIGHT= 6 TOP WEIGHT= 10
Configuration 1 Symmetry 1 110
Configuration 2 Symmetry 2 1ab
Configuration 3 Symmetry 1 101
Configuration 4 Symmetry 1 a1b
Configuration 5 Symmetry 2 ab1
Configuration 6 Symmetry 1 011
NO OF BASIS FUNCTIONS = 6 NO TO BE DELETED = 0
CI Matrix Elements calculated here
NO. OF CONFIGURATIONS IN REFERENCE SPACE = 1
SECONDARY SPACE = 6
TERTIARY SPACE = 6
NO. OF ORBITALS = 3
NO. OF ELECTRONS = 4
NO. OF WEIGHTS = 5
REFERENCE STATE CONFIGURATIONS ARE: 0
NO. OF CORE ORBITALS = 0
OPTION: NON-DIAGONAL HOLE LINE INTERACTIONS INCLUDED
Len28= 512 LenMCI= 241.
Leave Link 405 at Fri Feb 07 11:51:37 2025, MaxMem= 33554432 cpu: 1.0
(Enter C:\G09W\l510.exe)
Enter MCSCF program.
NO. OF ORBITALS = 74 NO. OF CORE-ORBITALS = 6
NO. OF VALENCE-ORBITALS = 3 NO. OF VIRTUAL-ORBITALS = 65
USED ACCURACY IN CHECKING CONVERGENCE = 1.00D-08
Memory needed for Incore Integrals: 7057107
Integrals KEPT IN MEMORY
IBUJAK length= 49980
Integral file not found: evaluate integrals
FoFCou: FMM=F IPFlag= 0 FMFlag= 0 FMFlg1= 0
NFxFlg= 0 DoJE=F BraDBF=F KetDBF=F FulRan=T
wScrn= 0.000000 ICntrl= 600 IOpCl= 0 I1Cent= 0 NGrid= 0
NMat0= 1 NMatS0= 1 NMatT0= 0 NMatD0= 1 NMtDS0= 0 NMtDT0= 0
Symmetry not used in FoFCou.
Defining IBUGAM
State Average Calculation. The weights are:
St.: 1 w.=0.333300 # St.: 2 w.=0.333300 # St.: 3 w.=0.333300 # St.:
2ND ORD PT ENERGY CV -0.004048 CU -0.048133 UV -0.040641
TOTAL -113.503472
ITN= 1 MaxIt= 64 E= -113.4106496306 DE=-1.13D+02 Acc= 1.00D-08 Lan= 0
ITN= 2 MaxIt= 64 E= -113.4944047782 DE=-8.38D-02 Acc= 1.00D-08 Lan= 0
ITN= 3 MaxIt= 64 E= -113.5020463877 DE=-7.64D-03 Acc= 1.00D-08 Lan= 0
ITN= 4 MaxIt= 64 E= -113.5089633540 DE=-6.92D-03 Acc= 1.00D-08 Lan= 0
ITN= 5 MaxIt= 64 E= -113.5068700003 DE= 2.09D-03 Acc= 1.00D-08 Lan= 0
ITN= 6 MaxIt= 64 E= -113.5081119793 DE=-1.24D-03 Acc= 1.00D-08 Lan= 0
ITN= 7 MaxIt= 64 E= -113.5074656605 DE= 6.46D-04 Acc= 1.00D-08 Lan= 0
ITN= 8 MaxIt= 64 E= -113.5076941123 DE=-2.28D-04 Acc= 1.00D-08 Lan= 0
ITN= 9 MaxIt= 64 E= -113.5075431541 DE= 1.51D-04 Acc= 1.00D-08 Lan= 0
ITN= 10 MaxIt= 64 E= -113.5075816620 DE=-3.85D-05 Acc= 1.00D-08 Lan= 0
ITN= 11 MaxIt= 64 E= -113.5075484014 DE= 3.33D-05 Acc= 1.00D-08 Lan= 0
ITN= 12 MaxIt= 64 E= -113.5075542665 DE=-5.87D-06 Acc= 1.00D-08 Lan= 0
ITN= 13 MaxIt= 64 E= -113.5075470663 DE= 7.20D-06 Acc= 1.00D-08 Lan= 0
ITN= 14 MaxIt= 64 E= -113.5075478623 DE=-7.96D-07 Acc= 1.00D-08 Lan= 0
ITN= 15 MaxIt= 64 E= -113.5075463471 DE= 1.52D-06 Acc= 1.00D-08 Lan= 0
ITN= 16 MaxIt= 64 E= -113.5075464600 DE=-1.13D-07 Acc= 1.00D-08 Lan= 0
ITN= 17 MaxIt= 64 E= -113.5075461684 DE= 2.92D-07 Acc= 1.00D-08 Lan= 0
ITN= 18 MaxIt= 64 E= -113.5075462023 DE=-3.38D-08 Acc= 1.00D-08 Lan= 0
ITN= 19 MaxIt= 64 E= -113.5075461614 DE= 4.09D-08 Acc= 1.00D-08 Lan= 0
ITN= 20 MaxIt= 64 E= -113.5075461812 DE=-1.98D-08 Acc= 1.00D-08 Lan= 0
ITN= 21 MaxIt= 64 E= -113.5075461839 DE=-2.73D-09 Acc= 1.00D-08 Lan= 0
... Do an extra-iteration for final printing.
EIGENVALUES AND EIGENVECTORS OF CI MATRIX
( 1) EIGENVALUE -113.9372546122
( 1) 0.8721303 ( 4)-0.4842478 ( 6)-0.0681868 ( 3)-0.0156029 ( 2) 0.0000000 ( 5) 0.0000000 (
( 2) EIGENVALUE -113.7834088173
( 2) 0.9999588 ( 5) 0.0090727 ( 4) 0.0000000 ( 1) 0.0000000 ( 3) 0.0000000 ( 6) 0.0000000 (
( 3) EIGENVALUE -113.5075461955
( 4) 0.8726403 ( 1) 0.4822660 ( 3) 0.0634586 ( 6)-0.0434911 ( 2) 0.0000000 ( 5) 0.0000000 (
Final one electron symbolic density matrix:
1 2 3
1 0.123472D+01
2 -0.284709D-12 0.199195D+01
3 -0.108298D+01 -0.182632D-12 0.773338D+00
MCSCF converged.
Leave Link 510 at Fri Feb 07 11:51:45 2025, MaxMem= 33554432 cpu: 8.0
(Enter C:\G09W\l601.exe)
Copying SCF densities to generalized density rwf, IOpCl= 0 IROHF=3.
**********************************************************************
Population analysis using the SCF density.
**********************************************************************
Orbital symmetries:
Occupied (A') (A') (A') (A') (A') (A') (A") (A')
Virtual (A") (A') (A') (A') (A") (A') (A') (A') (A') (A')
(A') (A") (A") (A') (A') (A") (A') (A') (A') (A')
(A") (A") (A') (A') (A') (A') (A") (A") (A') (A")
(A') (A') (A') (A') (A') (A") (A') (A') (A") (A")
(A') (A') (A") (A') (A") (A') (A") (A') (A') (A')
(A') (A') (A") (A') (A") (A') (A") (A') (A") (A')
(A') (A") (A') (A') (A') (A')
The electronic state is 1-A'.
Alpha occ. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha occ. eigenvalues -- 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000 0.00000 0.00000 0.00000 0.00000
Alpha virt. eigenvalues -- 0.00000
Condensed to atoms (all electrons):
1 2 3 4
1 C 4.810169 0.414962 0.414913 0.002124
2 H 0.414962 0.604744 -0.075572 -0.057680
3 H 0.414913 -0.075572 0.604694 -0.057628
4 O 0.002124 -0.057680 -0.057628 8.698157
Mulliken charges:
1
1 C 0.357833
2 H 0.113546
3 H 0.113593
4 O -0.584972
Sum of Mulliken charges = 0.00000
Mulliken charges with hydrogens summed into heavy atoms:
1
1 C 0.584972
4 O -0.584972
Electronic spatial extent (au): <R**2>= 61.1733
Charge= 0.0000 electrons
Dipole moment (field-independent basis, Debye):
X= -0.0001 Y= -3.2032 Z= 0.0000 Tot= 3.2032
Quadrupole moment (field-independent basis, Debye-Ang):
XX= -11.4588 YY= -13.8208 ZZ= -11.9322
XY= 0.0006 XZ= 0.0000 YZ= 0.0000
Traceless Quadrupole moment (field-independent basis, Debye-Ang):
XX= 0.9451 YY= -1.4169 ZZ= 0.4717
XY= 0.0006 XZ= 0.0000 YZ= 0.0000
Octapole moment (field-independent basis, Debye-Ang**2):
XXX= 0.0004 YYY= 0.3317 ZZZ= 0.0000 XYY= -0.0009
XXY= -0.4736 XXZ= 0.0000 XZZ= 0.0000 YZZ= 1.4023
YYZ= 0.0000 XYZ= 0.0000
Hexadecapole moment (field-independent basis, Debye-Ang**3):
XXXX= -17.5288 YYYY= -47.7035 ZZZZ= -12.4956 XXXY= -0.0001
XXXZ= 0.0000 YYYX= 0.0017 YYYZ= 0.0000 ZZZX= 0.0000
ZZZY= 0.0000 XXYY= -10.1164 XXZZ= -5.3572 YYZZ= -11.0727
#6 Quantum Chemistry » Question about units of first hyperpolarizability? » 2024-07-17 10:24:39
- choconostle123
- Replies: 1
Hello everyone i have a doub about the units of the firts hyperpolarizability, why the static first hyperpolarizability in some articles are reported in [10^-30 (cm^5)/esu] and in others are in [10^-48 esu]?
#7 Re: Quantum Chemistry » doubt about programing algorithm called "on grid method" » 2023-04-26 17:57:59
sorry is basin, I corrected it, oooo now I understand, thanks for your support i will train use a smaller grid spacing
#8 Quantum Chemistry » doubt about programing algorithm called "on grid method" » 2023-04-26 01:15:06
- choconostle123
- Replies: 2
hello everyone, i am learning to programing the algorithm called "on-grid method" to generate the basin, but i have a problem
i use the wave function of hydrogen molecule
my algorithm correctly assigns most of the points to the basin of the hydrogen 1 and hydrogen 2 (the bcp is at (0,0,0), and my nuclear attractors in (0,0,+-0.584)) but in the plane xy where is the bond critical point (points like (0,2,0) (2,2,0) (1.8,2,0)) these kind of points are assigned to the basis of hydrogen 1 so when i want to integrating the density, the density in the basin of hydrogen 1 has major density than basis of hydrogen 2 because if use 41 points of size in the end of my algorithm hydrogen1 has 36141 points in the basin and hydrogen2 34460
so i don't know wath to do for improving my algorithm ![]()
#9 Quantum Chemistry » A question about the linear response theory? » 2023-03-15 20:59:06
- choconostle123
- Replies: 0
I am studying linear response theory and i have a dub :S
if i have <Ψ(0)|O(interaction)|Ψ(0)> ¿can i say that is the same that? <Ψ(0)|O|Ψ(0)>
where Ψ(0) is the wave fuction in t=0 and O is and operator, O(interaction) is the operator in the interaction picture and O is the operator in the schrodinger picture
#10 Quantum Chemistry » Optimization with field » 2023-03-01 00:24:34
- choconostle123
- Replies: 1
Hello everyone, I have a problem with optimization with Gaussian with electric field
i want to get the wavefunction with the imput that is below but in the file .log don't converge the parameter maximum force:
Maximum Force 0.001404 0.000450 NO
RMS Force 0.000239 0.000300 YES
Maximum Displacement 0.001001 0.001800 YES
RMS Displacement 0.000265 0.001200 YES
i compute a optimization with AM1 and after that reoptimize with wb97x and finally i tried to carry on the optimization with electric field
can somebody help me?
Imput
######################################################################################
# wb97x/6-311g(d,p) opt=z-matrix
field=z+25 nosymm output=wfx
Title
0 1
C
C 1 B1
C 2 B2 1 A2
C 3 B3 2 A3 1 D3
C 4 B4 3 A4 2 D4
C 1 B5 2 A5 3 D5
H 1 B6 2 A6 3 D6
H 2 B7 1 A7 3 D7
H 4 B8 3 A8 2 D8
H 5 B9 4 A9 3 D9
N 6 B10 1 A10 2 D10
C 11 B11 6 A11 1 D11
H 12 B12 11 A12 6 D12
H 12 B13 11 A13 6 D13
H 12 B14 11 A14 6 D14
C 11 B15 6 A15 1 D15
H 16 B16 11 A16 6 D16
H 16 B17 11 A17 6 D17
H 16 B18 11 A18 6 D18
C 3 B19 2 A19 1 D19
C 20 B20 3 A20 2 D20
C 20 B21 3 A21 2 D21
C 21 B22 20 A22 3 D22
C 21 B23 20 A23 3 D23
N 2 B24 1 A24 3 D24
N 2 B25 1 A25 3 D25
N 2 B26 1 A26 3 D26
Variables:
B1 1.37560
B2 1.40539
A2 121.86975
B3 1.40508
A3 117.26966
D3 1.95844
B4 1.37577
A4 121.48013
D4 358.75293
B5 1.41364
A5 120.84104
D5 359.06738
B6 1.08201
A6 118.60060
D6 179.33656
B7 1.08556
A7 118.45003
D7 180.53095
B8 1.08248
A8 120.72238
D8 177.22259
B9 1.08205
A9 118.41558
D9 179.11626
B10 1.36166
A10 121.39303
D10 179.73340
B11 1.45061
A11 120.14828
D11 1.16008
B12 1.09642
A12 111.72966
D12 61.40646
B13 1.08892
A13 109.11361
D13 180.79604
B14 1.09567
A14 111.51731
D14 299.90691
B15 1.45093
A15 120.22106
D15 178.83967
B16 1.09547
A16 111.44904
D16 59.54934
B17 1.08887
A17 109.04696
D17 178.60271
B18 1.09642
A18 111.76132
D18 298.02621
B19 1.45906
A19 119.44899
D19 181.09297
B20 1.36427
A20 128.48543
D20 159.33459
B21 1.44441
A21 115.69955
D21 340.11136
B22 1.43375
A22 121.32581
D22 178.10742
B23 1.43167
A23 123.77609
D23 356.41187
B24 5.20007
A24 117.64799
D24 342.65979
B25 5.90532
A25 162.72252
D25 350.93398
B26 3.48548
A26 157.23346
D26 147.09305
#11 Re: Multiwfn and wavefunction analysis » dub TDM » 2022-11-27 01:06:26
Now I understand, thank you very much for the support and attention ![]()
#12 Re: Multiwfn and wavefunction analysis » dub TDM » 2022-11-26 17:58:51
Hello one doub, i have a question in the expresion of TDM between ground state and a selected excited state K in the section 3.21.9 of the manual
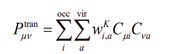
in this expression is need multyply by 2 ?
because for example i did a calculation with hydrogen molecule, the MO coefficients are

and the coeficient determinant is 0.70711
the elements of the TDM that result with the funtion 9 of excited state are

but the only way that i can get this results is for example the first with:
P_11=2*0.70711*0.548282599*1.21854048
#13 Multiwfn and wavefunction analysis » first hyperpolarizability and nonlinear absorption coefficient » 2022-06-14 17:21:06
- choconostle123
- Replies: 0
hello someone can i help with this question?
there is a mathematical way where can i relate the first hyperpolarizability result from gaussian with nonlinear absorption coefficient from the z-scan?
thanks for your attention
#14 Re: Multiwfn and wavefunction analysis » NTO projections on atomic functions » 2021-03-09 04:26:54
Hi
I did a calculation in gamess with CIS and get the wavefunction in .dat
$CONTRL SCFTYP=RHF CITYP=CIS AIMPAC=.TRUE. $END
$SYSTEM TIMLIM=3000 MEMORY=3000000 $END
$BASIS GBASIS=N311 NGAUSS=6 NDFUNC=1 NPFUNC=1 DIFFSP=.TRUE. DIFFS=.TRUE. $END
$CIS NSTATE=10 ISTATE=0 IROOT=0 $END
after that i converted the file .dat to wfn so, i want to convert the natural orbitals in canonical and my question is: can i convert natural orbitals in canonical orbitals ?
#15 Re: Multiwfn and wavefunction analysis » SOS computation of dipole hyperpolarizabilities » 2020-09-25 17:19:46
very grateful for the answer finally i can compare my values with the experimental values, and i want to comment a doubt in the meaning of the columns of the file beta_n because at difference with the file beta_n_comp in this file the meaning of each column appers but in the file beta_n don't appers i tried open with txt and opera (adjoint picture)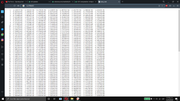

#16 Re: Multiwfn and wavefunction analysis » SOS computation of dipole hyperpolarizabilities » 2020-09-17 23:27:45
Hi, first i want to thank for the program in the last days this program and the manual has helped me a lot, and i want to ask about, which value can i use of my output of SOS to compare with the experimental value? in the paper are reported beta(zero) in 9 esu (they extrapolated the experimental value measure at 1580 nm), i have in my output of SOS of beta(0,0) the next data (adjoint picture), but i don't know which value i have to converted to esu and copare with the experimental value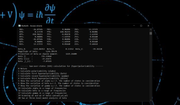
Pages: 1