Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Multiwfn and wavefunction analysis » ELF and LOL » 2021-06-05 12:14:26
- sourikanair
- Replies: 1
I wish to study localization of electrons in dexamethasone molecule. While executing ELF of the molecule in a plane, I saw lot of options such as XY plane 2: XZ plane 3: YZ plane 4: Define by three atoms etc. To plot the entire ELF of the molecule which option is suitable?
#2 Multiwfn and wavefunction analysis » NTO analysis error » 2020-07-17 05:08:59
- sourikanair
- Replies: 1
I did TDDFT calculations using ORCA and loaded .gbw file to run NTO analysis. Did calculation and got value of HOMO-LUMO energy.

But when I repeat the process again I am not getting energy gap. HOMO and LUMO have same energies
55(H ) --> Charge: 1.000000 x,y,z(Bohr): -11.214523 -3.650781 0.598930
56(H ) --> Charge: 1.000000 x,y,z(Bohr): -6.178762 1.802666 -4.288639
57(H ) --> Charge: 1.000000 x,y,z(Bohr): -0.057032 -5.519588 2.665837
Note: Orbital 105 is HOMO, energy: 0.978720 a.u. 26.632331 eV
Orbital 106 is LUMO, energy: 0.978720 a.u. 26.632331 eV
HOMO-LUMO gap: 0.000000 a.u. 0.000000 eV 0.000000 kJ/mol
What may be the reason?
#3 Re: Multiwfn and wavefunction analysis » Fukui calculations » 2020-07-16 07:01:29
Sir, I have calculated N.wfn,N+1.wfn and N-1.wfn.
Then used main function 100 and generated CDFT.txt.
output is ;ike this
Note: the E(HOMO) of TCE used for evaluating nucleophilicity index is the value evaluated at B3LYP/6-31G* level
Hirshfeld charges, condensed Fukui functions and condensed dual descriptors
Units used below are "e" (elementary charge)
Atom q(N) q(N+1) q(N-1) f- f+ f0 CDD
1(C ) -0.0139 -0.0139 -0.0139 0.0000 0.0000 0.0000 0.0000
2(C ) -0.0454 -0.0454 -0.0454 0.0000 0.0000 0.0000 0.0000
3(C ) 0.1229 0.1229 0.1229 0.0000 0.0000 0.0000 0.0000
4(C ) -0.0560 -0.0560 -0.0560 0.0000 0.0000 0.0000 0.0000
5(C ) 0.0269 0.0269 0.0269 0.0000 0.0000 0.0000 0.0000
6(C ) -0.0458 -0.0458 -0.0458 0.0000 0.0000 0.0000 0.0000
7(C ) -0.0465 -0.0465 -0.0465 0.0000 0.0000 0.0000 0.0000
8(C ) -0.0234 -0.0234 -0.0234 0.0000 0.0000 0.0000 0.0000
9(C ) -0.0247 -0.0247 -0.0247 0.0000 0.0000 0.0000 0.0000
10(C ) -0.0506 -0.0506 -0.0506 0.0000 0.0000 0.0000 0.0000
11(C ) -0.0175 -0.0175 -0.0175 0.0000 0.0000 0.0000 0.0000
12(C ) 0.0765 0.0765 0.0765 0.0000 0.0000 0.0000 0.0000
13(C ) 0.0168 0.0168 0.0168 0.0000 0.0000 0.0000 0.0000
14(C ) -0.0604 -0.0604 -0.0604 0.0000 0.0000 0.0000 0.0000
15(C ) 0.0495 0.0495 0.0495 0.0000 0.0000 0.0000 0.0000
16(C ) 0.1136 0.1136 0.1136 0.0000 0.0000 0.0000 0.0000
17(C ) 0.0149 0.0149 0.0149 0.0000 0.0000 0.0000 0.0000
18(O ) -0.2755 -0.2755 -0.2755 0.0000 0.0000 0.0000 0.0000
19(C ) -0.0839 -0.0839 -0.0839 0.0000 0.0000 0.0000 0.0000
20(C ) -0.0966 -0.0966 -0.0966 0.0000 0.0000 0.0000 0.0000
21(C ) -0.0842 -0.0842 -0.0842 0.0000 0.0000 0.0000 0.0000
22(C ) 0.1545 0.1545 0.1545 0.0000 0.0000 0.0000 0.0000
23(O ) -0.2113 -0.2113 -0.2113 0.0000 0.0000 0.0000 0.0000
24(C ) 0.0103 0.0103 0.0103 0.0000 0.0000 0.0000 0.0000
25(O ) -0.2373 -0.2373 -0.2373 0.0000 0.0000 0.0000 0.0000
26(O ) -0.1886 -0.1886 -0.1886 0.0000 0.0000 0.0000 0.0000
27(O ) -0.1865 -0.1865 -0.1865 0.0000 0.0000 0.0000 0.0000
#4 Re: Multiwfn and wavefunction analysis » Fukui calculations » 2020-07-14 15:59:07
Thank you Tian Lu
#5 Re: Multiwfn and wavefunction analysis » Fukui calculations » 2020-07-14 11:30:28
Sir,
The three states generated are .gjf file. How can we convert them to .gbw? I think ORCA won't use .gjf file as input
#6 Multiwfn and wavefunction analysis » Fukui calculations » 2020-07-14 05:36:14
- sourikanair
- Replies: 6
I want to calculate fukui functions without Gaussian. I use Orca. How can I generate N.wfn ,(N+1).wfn and (N-1).wfn from .gjf.
I think Orca cannot input .gjf file
#7 Re: Multiwfn and wavefunction analysis » Bat file not executing » 2020-07-09 16:47:02
I have done all that correct. Again same output. WHat might be the reason? Is there any alternative method
#8 Multiwfn and wavefunction analysis » Bat file not executing » 2020-07-09 11:06:00
- sourikanair
- Replies: 3
While AIM analysis and ALIE analysis my .bat file is not working. Tried a lot.
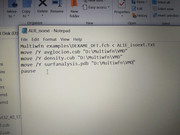
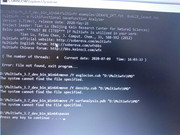
But couldn't identify the problem. Not sure if there is any problem in editing bat file. Hence I am attaching the details of bat file and output that i had got. Please help me resolving this issue
#9 Multiwfn and wavefunction analysis » AIM topology map in VMD » 2020-05-27 14:20:57
- sourikanair
- Replies: 1
Copied AIM.bat and AIM.txt from "examples\scripts" to the executable file. Edited ip in the AIM.bat, modified the default VMD folder to actual VMD folder. but couldn't run the batch file. showing error. couldnt generate pdb files
Pages: 1