Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » Skewness » 2023-12-25 03:43:54
Thanks!
#2 Multiwfn and wavefunction analysis » Skewness » 2023-12-24 14:12:18
- Ossama
- Replies: 2
Dear Professor Tian Lu
In the "Quantitative analysis of molecular surface", is it possible to add the skewness of the investigated function (including, but not limited to, positive and negative electrostatic potentials)?
The definition of skewness which I am working on is displayed in the attached image.
Of course I can export the surface vertices, and calculate any statistical parameter, but this would be tiresome for a large number of molecules. I also think that skewness may be a good addition to Multiwfn Software.
Thanks and regards,,

#3 Re: Multiwfn and wavefunction analysis » ELF as user-defined function » 2023-02-22 02:31:30
Thank you so much!
#4 Multiwfn and wavefunction analysis » ELF as user-defined function » 2023-02-21 15:49:43
- Ossama
- Replies: 6
Hello!
In "quantitative analysis of molecular surface", is there a way to make the electron localization function (ELF) the user-defined function?
In the manual, there are over 100 functions of which none is ELF.
I know there are many ready-to-use user-defined functions that are pertinent to Pauli repulsion, but I wished if ELF specifically was available. I also am aware that I can export cubes of the electron density and ELF then map the later on the electron density isosurface and perform the molecular surface analysis, but for large numbers of molecules this consumes as much as 10 times effort.
Thanks!
#5 Re: Multiwfn and wavefunction analysis » GAMESS optimization .out files » 2023-02-04 05:56:49
Do you want to let Multiwfn automatically load the wavefunction at optimized geometry?
Yes.
If yes, perhaps this cannot be realized, because I found GAMESS doesn't print expansion coefficients of all orbitals for final geometry. If you have a way to make GAMESS output all orbital coefficients of final geometry in output file, I can modify Multiwfn to load them instead of loading the EIGENVECTORS of the initial geometry.
Well. Thanks!
I will check GAMESS documentation and ask for help in GAMESS google groups. In case a solution is found, I will make an update in this thread to inform you, Professor.
Meanwhile, I will keep using the usual method, i.e., carrying out another GAMESS job using the optimized geometry then loading the output into Multiwfn as input.
#6 Multiwfn and wavefunction analysis » GAMESS optimization .out files » 2023-02-02 21:47:29
- Ossama
- Replies: 4
Hi!
I use GAMESS for geometry optimization and get .out files. In order to use Multiwfn, I have to rerun single point energy calculation for the new optimized geometry to use the new .out files as input into Multiwfn (after changing the extension to .gms, of course)
Whenever I use the optimization .out files in Multiwfn I get this message:
"Warning: This is an optimization task, only wavefunction corresponding to initial geometry will be loaded
Press ENTER button to continue"
Is there a way to get around this problem?
Thanks,
#7 Re: Multiwfn and wavefunction analysis » Electric dipole moment » 2022-08-07 22:34:08
Thanks, Prof. Lu
It worked.
I downloaded the latest version (3.8) since the function you mentioned didn't exist in the previous versions. Both files of Cl2 gave a dipole moment magnitude of zero, which, of course, is the expected value for a homonuclear diatomic molecule.
My regards,
#8 Re: Multiwfn and wavefunction analysis » Electric dipole moment » 2022-08-07 12:32:49
I tried to modify the X, Y and Z components of the electric dipole moments by adding the effect of the two chlorines' nuclei (for both files) but I failed to get identical values of the total magnitude.
#9 Re: Multiwfn and wavefunction analysis » Electric dipole moment » 2022-08-07 12:24:25
Dear Prof. Lu,
As mentioned in Multiwfn manual (3.6- Outputting and plotting specific property within a spatial region):
"If what you calculated is electron density, the molecular dipole moment evaluated based on grid data is also printed out."
I carry out the following steps:
5 Output and plot specific property within a spatial region (calc. grid data)
1 Electron density
3 High quality grid , covering whole system, about 1728000 points in total
I've an example here, Cl2 molecule, where two .wfn files were used. There is no difference between them other than the coordinates origin.
The first one (Cl2.wfn) gives:
Electric dipole moment estimated by integrating electron density
X component: 4.596854 a.u. 11.684035 Debye
Y component: -1.037455 a.u. -2.636946 Debye
Z component: -0.035310 a.u. -0.089750 Debye
Total magnitude: 4.712602 a.u. 11.978239 Debye
The second one (Cl2-reoriented.wfn) gives:
Electric dipole moment estimated by integrating electron density
X component: -0.218321 a.u. -0.554918 Debye
Y component: -0.386888 a.u. -0.983370 Debye
Z component: -0.035310 a.u. -0.089750 Debye
Total magnitude: 0.445638 a.u. 1.132699 Debye
#10 Multiwfn and wavefunction analysis » Electric dipole moment » 2022-08-06 14:54:01
- Ossama
- Replies: 5
Hi
Forgive me if this is a naïve question!
I find that the electric dipole moment of a molecule, calculated after the grid of electron density is calculated, is dependent on the origin coordinate. This makes the obtained result rather meaningless. I suppose if the nuclear charges were incorporated in the calculations this would make the obtained result origin-independent, at least for neutral molecules or a zero-net charge distribution.
1- Am I right?
2- And if so, is there a handy way to calculate the electric dipole moment without excluding the nuclear charges?
Thanks,,
#11 Re: Multiwfn and wavefunction analysis » Oxygen (O2) with restricted and unrestricted wavefunctions » 2022-07-30 21:48:07
Update:
I've tried UHF method with N2 and, using Multiwfn, it gave identical results to those of RHF method. So I assumed there was no problem with UHF per se. I tried UHF method again with O2 but with multiplicity = 1 rather than 3, and it gave identical results to those of RHF method (with multiplicity = 1, of course).
I concluded that it was all about multiplicity not about the nature of the Hartree-Fock method. So I think the results mentioned in the original post, indeed, seem to be correct and that the O2 case is different than N2 and F2 due to its different multiplicity.
#12 Re: Multiwfn and wavefunction analysis » Oxygen (O2) with restricted and unrestricted wavefunctions » 2022-07-30 15:01:51
I have checked O2-UHF.wfn, the result seems to be correct. ESP is indeed positive everywhere on the vdW surface. This is never necessarily wrong.
Well. At least this means there is nothing wrong with my input file.
I'am working on a series of diatomic molecules. N2 and F2 molecules exhibit areas with positive and negative values. Additionally, O2 with RHF-based wavefunction is consistent with that trend. I cannot think of an intuitive reason that makes oxygen different.
BTW: It can be proven that for isolated atoms, ESP is also positive everywhere in the whole space.
Yes it can, due to the concentration of all the positive charge in one point while the counterpart negative charge is distributed over a relatively large space. But my intuition, along with some calculations of course, tells me that for diatomic molecules, whether homonuclear or heteronuclear, we should obtain both positive and negative areas on VdW surface. I will reinvestigate the O2 case anyway.
Thanks and regards,
#13 Multiwfn and wavefunction analysis » Oxygen (O2) with restricted and unrestricted wavefunctions » 2022-07-30 05:05:27
- Ossama
- Replies: 3
Hello!
When I perform a molecular surface analysis of oxygen (O2), using GAMESS-US .wfn file (extracted from the .dat file) as input, the obtained results depend on whether the Hartree-Fock wavefunction is restricted or unrestricted.
The problem is that with the unrestricted wavefunction the results are not chemically sound. For example, all the molecular surface has a positive ESP:
,,
Minimal value: 0.10957 kcal/mol Maximal value: 3.56581 kcal/mol
Overall surface area: 179.61505 Bohr^2 ( 50.29734 Angstrom^2)
Positive surface area: 179.61505 Bohr^2 ( 50.29734 Angstrom^2)
Negative surface area: 0.00000 Bohr^2 ( 0.00000 Angstrom^2)
,,
The restricted HF method gives sound results in Multiwfn analysis, but it, simply, is not the best practice. Should I use the restricted HF instead of UHF in Multiwfn analysis?
#14 Re: Multiwfn and wavefunction analysis » Unrealistic MEP analysis with GAMESS-US wfn files » 2022-07-29 14:14:37
Thanks for you time!
I've corrected it and it worked.
#15 Multiwfn and wavefunction analysis » Unrealistic MEP analysis with GAMESS-US wfn files » 2022-07-29 07:39:06
- Ossama
- Replies: 2
Hello!
I am performing a "quantitative analysis of molecular surface" for some molecules like, for example, F2, Cl2, Br2, I2.
I use GAMESS software where I take the last section of .dat files that begins with "----- TOP OF INPUT FILE FOR BADER'S AIMPAC PROGRAM -----" and create my own wfn files.
My problem occurs when I use effective core potentials for relatively large atoms like Br and I. In this case I get unreasonable results where all the molecular surface has a positive electrostatic potential value.
I get the effective core potentials from the basis-set exchange site: https://www.basissetexchange.org/. When I was working with Gaussian09, it was okay and the results were sound, but this is the first time where I use GAMESS with explicit basis sets.
For instance, for iodine (I2), I get these results from Multiwfn 3.7 (GAMESS input and output files are attached at the end of the post):
Volume: 710.01669 Bohr^3 ( 105.21364 Angstrom^3)
Estimated density according to mass and volume (M/V): 4.0058 g/cm^3
Minimal value: 5880.60843 kcal/mol Maximal value: 7195.40597 kcal/mol
Overall surface area: 407.16839 Bohr^2 ( 114.01878 Angstrom^2)
Positive surface area: 407.16839 Bohr^2 ( 114.01878 Angstrom^2)
Negative surface area: 0.00000 Bohr^2 ( 0.00000 Angstrom^2)
Overall average value: 9.99750281 a.u. ( 6273.53299 kcal/mol)
Positive average value: 9.99750281 a.u. ( 6273.53299 kcal/mol)
Negative average value: NaN a.u. ( NaN kcal/mol)
Overall variance (sigma^2_tot): 0.43247968 a.u.^2 (170297.00593 (kcal/mol)^2)
Positive variance: 0.43247968 a.u.^2 ( 170297.00593 (kcal/mol)^2)
Negative variance: 0.00000000 a.u.^2 ( 0.00000 (kcal/mol)^2)
Balance of charges (nu): 0.00000000
Product of sigma^2_tot and nu: 0.00000000 a.u.^2 ( 0.00000 (kcal/mol)^2)
Internal charge separation (Pi): 0.55515652 a.u. ( 348.36627 kcal/mol)
Molecular polarity index: 272.04588592 eV ( 6273.53299 kcal/mol)
Nonpolar surface area (|ESP| <= 10 kcal/mol): 0.00 Angstrom^2 ( 0.00 %)
Polar surface area (|ESP| > 10 kcal/mol): 114.02 Angstrom^2 (100.00 %)
The GAMESS input and output (extracted wfn file) are attached to these links:
#16 Re: Multiwfn and wavefunction analysis » Vomume enclosed by a function isosurface » 2020-05-18 08:08:47
What if I need to study the outer surface separately, its area, the volume which it encloses (all the enclosed volume), mapping it with electron density or electrostatic potential and finding out the areas of positive and negative electrostatic potential etc? Is it possible with Multiwfn? If I use Chimera, do I get what I need?
#17 Re: Multiwfn and wavefunction analysis » Vomume enclosed by a function isosurface » 2020-05-17 21:47:34
Regarding the second question, it is better to upload an image, otherwise I am unable to clearly understand your representation.
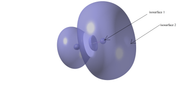
This is an ELF representation of FI molecule. The isosurface value is 0.2.
According to the ELF there are one (or more than one) isosurface that is contained in another isosurface. Here isosurface 2 encompasses isosurface 1, albeit being with the same value.
So when I am given the isosurface characteristics by Multiwfn like area, volume etc, does it mean the outer surface only or all the surfaces?
#18 Multiwfn and wavefunction analysis » Vomume enclosed by a function isosurface » 2020-05-17 19:29:01
- Ossama
- Replies: 5
Hi
Thank you for your effort. I am really impressed.
I've a question regarding defining an isosurface of the molecule in Multiwfn, in particular, the area of the surface or the volume enclosed by it.
In "Quantitative analysis of molecular sufrace", If I choose, say, electron localization function, and I set the subsurface value to be 0.7. Here I will have many isosurfaces that enclose many volumes. Then, the values of area and volume I get from Multiwfn, are they supposed to be the sum of all obtained isosurfaces' areas and volumes?
Besides, if I choose the isosurface value to be, say, 0.1. Here, I am supposed to have an isosurface that encloses other isosurfaces. Then, do the values obtained for the molecular surface and the volume enclosed represent the outer isosurface only?
Thanks!
Pages: 1