Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 Re: Multiwfn and wavefunction analysis » Interpretation of hole-electron analysis results using ChatGPT » 2025-07-30 06:27:26
Thank you for your response. I understand your position regarding AI-generated content. My intention was to validate and discuss the scientific interpretation, not the tool itself. I appreciate your time either way.
#2 Multiwfn and wavefunction analysis » Interpretation of hole-electron analysis results using ChatGPT » 2025-07-29 17:29:27
- may01dz
- Replies: 2
I wrote the prompt below in ChatGPT:
So I want to determine whether transition number 1 is a π–π*, σ–π*, or n–π* transition using the hole-electron analysis framework described in the attached paper ( "Liu et al. - 2020 - An sp-hybridized all-carboatomic ring, cyclo[18]carbon Electronic structure, electronic spectrum, a.pdf").
I used the Multiwfn program to obtain the parameters needed to identify the nature of this transition, which are shown in the table below.
Integral of hole: 0.960000
Integral of electron: 0.959942
Integral of transition density: 0.000039
Transition dipole moment in X/Y/Z: 0.428149 2.521187 -2.158261 a.u.
Sm index (integral of Sm function): 0.45245 a.u.
Sr index (integral of Sr function): 0.69978 a.u.
Centroid of hole in X/Y/Z: 7.818786 0.960151 2.729048 Angstrom
Centroid of electron in X/Y/Z: 7.534434 0.737712 2.573506 Angstrom
D_x: 0.284 D_y: 0.222 D_z: 0.156 D index: 0.393 Angstrom
Variation of dipole moment with respect to ground state:
X: 0.515837 Y: 0.403521 Z: 0.282165 Norm: 0.713116 a.u.
RMSD of hole in X/Y/Z: 1.365 1.709 2.496 Norm: 3.319 Angstrom
RMSD of electron in X/Y/Z: 1.173 1.932 2.114 Norm: 3.095 Angstrom
Difference between RMSD of hole and electron (delta sigma):
X: -0.191 Y: 0.223 Z: -0.382 Overall: -0.224 Angstrom
H_x: 1.269 H_y: 1.821 H_z: 2.305 H_CT: 1.654 H index: 3.207 Angstrom
t index: -1.261 Angstrom
Hole delocalization index (HDI): 7.27
Electron delocalization index (EDI): 7.41
Ghost-hunter index: -33.291 eV, 1st term: 3.340 eV, 2nd term: 36.631 eV
Excitation energy of this state: 3.091 eV
Based on the attached paper and the data in the results table, are there clear indicators of the nature of transition 1?
Note that transition 1 is characterized by the following:
STATE 1: E= 0.113590 au 3.091 eV 24930.0 cm**-1 <S**2> = 0.000000 Mult 1
74a -> 75a : 0.959961 (c= 0.97977605)
And the response was as follows.
Based on the data you provided and the criteria described in Liu et al. (2020) for hole-electron analysis, we can now interpret the nature of transition 1 (0–1A → 1–1A) using quantitative descriptors:
Key Descriptors from Your Multiwfn Output:
Integral of hole / electron
≈ 0.96
Very good quality, near-conservation of charge.
Integral of transition density
0.000039
Very small ⇒ transition is not charge-transfer (CT).
D index (distance between hole and electron centroids)
0.393 Å
Short ⇒ local excitation.
Sm index
0.452
Indicates strong spatial overlap.
Sr index
0.699
Also consistent with non-CT excitation.
Δσ (delta RMSD)
-0.224 Å
Small, no major shape difference.
Hole & Electron Delocalization Indices (HDI/EDI)
~7.3 / 7.4
Strongly delocalized ⇒ characteristic of π orbitals.
t index
–1.261 Å
Negative ⇒ not typical charge-transfer behavior.
Transition dipole vector norm
3.31 a.u.
Large, consistent with intense π–π* transition.
Excitation energy
3.091 eV (401.1 nm)
In the visible–near UV range.
Dominant configuration
74a → 75a with coefficient ≈ 0.98
Clean single excitation between adjacent orbitals.
Interpretation Using Hole-Electron Analysis Framework:
Liu et al. emphasize that π–π* transitions:
Occur with strong overlap between hole and electron regions (high Sm, moderate-to-high Sr).
Exhibit short D index and delocalized densities.
Show large oscillator strengths and transition dipole magnitudes due to strong spatial overlap.
Involve delocalized MOs such as πin or πout in conjugated systems.
By contrast:
n–π* transitions generally show more localized hole, small HDI, larger D and smaller oscillator strength.
σ–π* transitions usually originate from localized σ orbitals with small delocalization and different energy levels.
Conclusion:
All numerical evidence (short D, large Sm, strong delocalization indices, clean HOMO→LUMO excitation, high transition dipole moment) points toward:
Transition 1 is a strongly allowed π–π* excitation.
There is no indication of n or σ orbital involvement (e.g., low HDI, large D, weak overlap, or low fosc), which further excludes n–π* or σ–π* character.
How accurate were its answers and interpretations of the results, and can I rely on them?
#3 Re: Multiwfn and wavefunction analysis » Hirshfeld Surface Analysis of Zwitterionic Forms in a Schiff Base » 2025-07-21 21:47:30
Thank you very much for your helpful and detailed response. I truly appreciate your guidance and the references you shared. I plan to follow your advice and will proceed with the IRI analysis in Multiwfn to investigate the presence of the intramolecular hydrogen bond and assess the zwitterionic nature of the molecule.
#4 Multiwfn and wavefunction analysis » Hirshfeld Surface Analysis of Zwitterionic Forms in a Schiff Base » 2025-07-21 16:18:35
- may01dz
- Replies: 2
Hello,
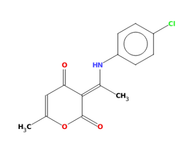
I would like to use Hirshfeld surface analysis in Multiwfn to help confirm or rule out the presence of zwitterionic forms in a Schiff base molecule (see attached image). Specifically, I aim to determine whether an intramolecular N⁺−H⋯O⁻ hydrogen bond exists, which would support the zwitterionic character of the compound.
#5 Re: Multiwfn and wavefunction analysis » Request for Workshops from Multiwfn Forum » 2024-03-22 21:22:45
Once again, thank you very much for your kind consideration and willingness to assist.
#6 Multiwfn and wavefunction analysis » Request for Workshops from Multiwfn Forum » 2024-03-19 04:48:09
- may01dz
- Replies: 2
Dear Mr. Tian Lu,
I am writing to express my admiration for the remarkable work being done on the Multiwfn forum in Chinese. It's evident that the forum is flourishing with an abundance of valuable information and engaging activities. One aspect that particularly stands out is the workshops you organize for Chinese students periodically.
I would like to inquire if there are any intentions or plans to organize workshops for a wider audience beyond the current scope. Your insights and teachings would be invaluable to us, and we are in dire need of such opportunities for growth and development.
Warm regards,
Mr. A.may
#7 Re: Multiwfn and wavefunction analysis » Activation Strain Model » 2022-09-13 22:19:38
thanks rikaaardoss, it's helpful
Hello!, sorry to intrude, but no specific software is required to perform ASM calculations. It is only necessary to calculate the distortion energy (or also called preparation energy) and the interaction energy.
The former is calculated from the difference of the formed complex (independent of whether it is a local minimum or a transition state) and the free (i.e. optimized) fragments. Then, the interaction energy can be calculated through the difference of the energy of the complex and the fragments in the geometries they have in the complex (this is done through an SP calculation).
The ASM is also called the distortion/interaction model and was proposed by K. Houk, the studies of Houk's group emphasize mostly on the distortion energy, while bickelhaupt and the proponents of the ASM give more importance to the interaction energy and the energy decomposition analysis. But strictly speaking, that is already more EDA than ASM (or D-I model). Personally, what I have done with multiwfn by way of exploration is to perform a simple-EDA, however sometimes this analysis is somewhat incomplete or cumbersome to calculate (due to the need to calculate dispersion energies).
One suggestion for Multiwfn developers would be to be able to calculate the distortion and interaction energies through the IRC, I believe this would be relatively simple to implement, my programming skills are quite limited, however, I would like to be able to participate in the implementation through the application in a number of systems of interest.
Best regards and many thanks to developers
R.
#8 Multiwfn and wavefunction analysis » Activation Strain Model » 2022-09-08 15:57:16
- may01dz
- Replies: 4
Hi,
Does the "Activation Strain Model" Method exist in Multiwfn, or does it have one similar to it?
#9 Re: Multiwfn and wavefunction analysis » DCT indices » 2022-03-26 13:42:50
OK, thank you very much
#10 Re: Multiwfn and wavefunction analysis » DCT indices » 2022-03-26 09:04:59
thanks...but, it is D index in the Multiwfn screen output?
This index can be calculated both via hole-electron analysis and based on grid data of electron density difference (between ground and excited states). For the former case, see Section 4.18.1 of Multiwfn manual for example, for the latter case, see Section 4.18.3. The former is more convenient and usually more recommended.
#11 Multiwfn and wavefunction analysis » DCT indices » 2022-03-26 08:18:29
- may01dz
- Replies: 4
Hello.
How can I use the multiwfn program to calculate the DCT index?
Thank you so much
#12 Re: Multiwfn and wavefunction analysis » solubility with GIPF descriptors? » 2022-01-16 17:48:59
OK, thanks a lot
#13 Re: Multiwfn and wavefunction analysis » solubility with GIPF descriptors? » 2022-01-15 14:31:04
well,
1/Do you have a study or reference dealing with this issue? (Organometallic solubility)?
2/Other than a G09 output file and the Multiwfn programme, what do I need to get the solubility value?
3/Is there a similarity between GIPF and COSMO-RS?
GIPF descriptors are suitable for any kind of system.
#14 Multiwfn and wavefunction analysis » solubility with GIPF descriptors? » 2022-01-14 20:05:00
- may01dz
- Replies: 4
Hello,
Is it possible for GIPF descriptors to determine the solubility of a system containing metal atoms, such as Cu atom?
Thank you very much.
#15 Re: Quantum Chemistry » small basis sets for geometry optimization » 2021-10-10 20:51:49
OK, thank you very much.
This is a very common and widely accepted practice, see my paper Computational and Theoretical Chemistry, 1200, 113249 (2021) DOI: 10.1016/j.comptc.2021.113249 for example, in the examples in Section 4 I used a relatively low level for geometry optimization while significantly better level for energy evaluation.
#16 Quantum Chemistry » small basis sets for geometry optimization » 2021-10-09 19:49:37
- may01dz
- Replies: 2
Hello,
I was aware that in a geometry optimization procedure, you encourage the usage of small basis sets. Are there any papers that support this suggestion...? Thank you so very much.
#17 Re: Quantum Chemistry » imposing a point group symmetry » 2021-09-09 17:29:08
Thank you so much for your response, and please accept my apologies for the delay in responding because I only saw it now... In any case, thanks a lot
Hello,
the vmd program will do that for you as well in a very user friendly graphical environment. You will find the feature from the vmd main window under Extensions -> Analysis -> Symmetry Tool.
Regards,
Michael
#18 Re: Multiwfn and wavefunction analysis » ETS-NOCV has been supported in Multiwfn! » 2021-07-27 21:55:54
AHHHH, My thanks and appreciation
#19 Re: Multiwfn and wavefunction analysis » aromaticity index for Porphyrin-like systems » 2021-05-27 18:44:12
Hello,
The following are the responses I received from the Orca Forum's leaders:
For the second question (ORCA outputted only the shielding tensor of the last real atom (94H) and has not shown up that of 95H (the ghost atom).) the answer is as follows:
One of the many idiosyncrasies of the EPRNMR module is that counting for the NUCLEI keyword starts at 1, unlike most of the program. This is noted in the manual. So 94H is the 95th atom in the molecule. You can ask for atom 96, but it's probably easiest to just let the program calculate all shieldings - as discussed, the calculation time is basically the same.
As for the basis set on the ghost atom: it is not wrong per se to use a regular basis like def2-TZVP - indeed, it gets the globally assigned basis for hydrogen (or whatever element the ghost atom is). However, it does introduce a basis set error if you compare multiple calculations which only differ in the position of the ghost atom (or lack thereof). Since def2-TZVP is probably not at the CBS limit for shieldings, this error may be significant.
In other words, the basis set of the ghost atom is added to the basis set of the system, but what you probably want is to probe different spatial positions of the system with its basis set unchanged. You can do that with a dummy atom (X) but then the problem is that the latter has no grid points, so for DFT at points just a couple of Angstrom away from real atoms, you get totally wrong results. Hence, the workaround of using ghost atoms (which do get grid points) but giving them a practically non-interacting basis of a single tight s-function (and corresponding aux functions, if necessary).
the answer on - although I asked the program to calculate the magnetic shielding tensor of one atom, the time spend to do that was not less than that it spend to calculate the magnetic shielding tensor of all the atoms of the molecule, Does this make sense?- the respond was:
Most of the time is spent in the CPSCF equations that are done for the x,y and z component of the magnetic field.
The shielding tensor is a second derivative, so one could formulate it to first take the derivative with respect to the nuclear magnetic moment and then wrt to the magnetic field. Then you would benefit greatly in performance if you have only one nucleus (see work by Ochsenfeld et al who report this "sublinear scaling") but as soon as you would have more than just a few nuclei (as you would typically for 1H NMR) you would need much (!) more time. This is why most codes first do the field derivative, and also why you hardly save any time if you only need few nuclei as the second derivative is comparably cheap after the CPSCF equations have been done.
#20 Re: Multiwfn and wavefunction analysis » aromaticity index for Porphyrin-like systems » 2021-05-26 06:53:43
well, thank you very much
I'll post my inquiry in the ORCA forum, then I'll share the reply here (If you don't mind).
I am not familiar with this feature of ORCA. What I can say is that multicenter bond order (also known as multi-center bond index) and AV1245 in Multiwfn are suitable for investigating aromaticity of porphyrin-like systems. The ICSS supported by Multiwfn is very useful in visually studying aromaticity of this system, though the cost is high.
#21 Multiwfn and wavefunction analysis » aromaticity index for Porphyrin-like systems » 2021-05-25 17:57:13
- may01dz
- Replies: 3
Hello,
1/Which aromaticity index is appropriate for Porphyrin-like systems?
2/
I used ORCA and the corresponding input below to calculate the magnetic shielding tensor at a ghost atom H: 1 A° above the ring plan (-0.0676346525,0.0191628382,-0.8365329255). the order of the ghost atom is 95.
the structure of the input is:
%pal nprocs 10 end
%maxcore 3675
! B3LYP def2-TZVP def2/J def2/JK TightSCF NMR RijCosX GridX4
* xyz 0 1
C -2.82353463539941 -1.13724734028106 0.44846883140457
C -4.18974744461116 -0.69584407152261 0.53629948317338
.
.
.
H: -0.0676346525 0.0191628382 -0.8365329255 # This H atom is defined as a ghost atom
*
%EPRNMR
NUCLEI = 95 {SHIFT}
END
Somehow, ORCA outputted only the shielding tensor of the last real atom (94H) and has not shown up that of 95H (the ghost atom).
On the other hand, although I asked the program to calculate the magnetic shielding tensor of one atom, the time spend to do that was not less than that it spend to calculate the magnetic shielding tensor of all the atoms of the molecule, Does this make sense?
thank you very much
#22 Re: Multiwfn and wavefunction analysis » Error: Data needed by this function is not presented » 2021-05-11 14:17:59
OK, thanks
#23 Multiwfn and wavefunction analysis » Error: Data needed by this function is not presented » 2021-05-11 09:59:20
- may01dz
- Replies: 2
Hello,
I’m using the ORCA output as an input for Multiwfn, and, when I want to view the Molecular structure and the orbitals ( option 0), I got the following message error: “Error: Data needed by this function is not presented! Check your input file!”
Which keywords should I add to my ORCA'input file to overcome this issue?
thank you very much.
#24 Re: Quantum Chemistry » TD DFT emission spectrum. » 2021-03-31 20:13:13
Hello,
Follow the steps in the following reference:
#25 Re: Quantum Chemistry » imposing a point group symmetry » 2021-03-10 22:21:10
thanks, I will
Have a look at SYVA code, which is freely available and can be downloaded at Computer Physics Communications 215 (2017) 156–164. SYVA is able to detect point group and can present symmetrized structure.
#26 Re: Quantum Chemistry » imposing a point group symmetry » 2021-03-08 13:12:11
Hi,
I want to symmetrize it
Do you want to symmetrize your molecular structure? or simply detecting point group?
#27 Quantum Chemistry » imposing a point group symmetry » 2021-03-06 21:03:53
- may01dz
- Replies: 6
Hello,
I look for a free software, capable of imposing a point group symmetry to my molecule.
Thanks in advance.
#28 Re: Multiwfn and wavefunction analysis » Multiwfn is able to easily perform NCI analysis for periodic system » 2021-02-27 20:40:20
Hello,
If necessary ... we will learn Chinese.
#29 Re: Multiwfn and wavefunction analysis » AICD 2.0 with ORCA » 2021-02-18 16:21:44
Ah!!! I thought, Really, that it was one of your useful programs... Anyway, thank you very much.
It is impossible.
This problem is irrelevant to Multiwfn, you should ask developer of AICD code.
#30 Multiwfn and wavefunction analysis » AICD 2.0 with ORCA » 2021-02-17 12:53:21
- may01dz
- Replies: 2
Hello, Multiwfn users,
its possible for AICD to read the output of ORCA NMR calculation ?
thank you very much