Multiwfn forum
Multiwfn official website: http://sobereva.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Multiwfn and wavefunction analysis » Integrating with mixed type grid » 2026-05-27 15:54:55
- Alexey
- Replies: 1
Dear Tian Lu.
I'm faced with the task of integrating existing Bader basins, but in a specific way. The integration needs to be done in a mixed-type grid. For my task, I need to take only those voxels in which the value of my function (below, MYFUNC) is >= 0.5. Am I correct in understanding that I need to add code in two places in the basin.f90 file, namely:
1)
...
!Use DFT integration algorithm to integrate the region inside the trust radius
...
!$OMP do schedule(DYNAMIC)
do ipt=1,nintgrid
ptx=gridatt(ipt)%x
pty=gridatt(ipt)%y
ptz=gridatt(ipt)%z
MYFUNC = f(ptx, pty, ptz, "f") #my function
if (MYFUNC < 0.5D0) cycle
rx=ptx-CPpos(1,numcp) !The relative distance between the current point and the corresponding attractor
ry=pty-CPpos(2,numcp)
rz=ptz-CPpos(3,numcp)
!Calculate switching function
dist=dsqrt(rx*rx+ry*ry+rz*rz)
....
2)
...
!--------- Integrating uniform grids, basin boundary grids will be calculated in the later stage
!$OMP do schedule(DYNAMIC)
do iz=izlow,izup
do iy=iylow,iyup
do ix=ixlow,ixup
if ((itype==2.or.itype==3).and.interbasgrid(ix,iy,iz)) cycle !If refine boundary grid at next stage, we don't calculate them at present stage
call getgridxyz(ix,iy,iz,rnowx,rnowy,rnowz)
MYFUNC = f(rnowx, rnowy, rnowz, "f")
if (MYFUNC < 0.5D0) cycle
iatt=gridbas(ix,iy,iz)
! do icp=1,numcp
! dist=dsqrt( (rnowx-CPpos(1,icp))**2+(rnowy-CPpos(2,icp))**2+(rnowz-CPpos(3,icp))**2 )
! if (disttest<trustrad(icp)) cycle cycix !The function inside trust radius is integrated by DFT integration
! end do
rx=rnowx-CPpos(1,iatt) !The relative distance between the current point and the exact position of the corresponding attractor
ry=rnowy-CPpos(2,iatt)
...
I would be very grateful if you could tell me if I'm on the right track and if I need to add an "if MYFUNC..." condition somewhere else in the code (When integrating the basin boundary mb?)
#2 Re: Multiwfn and wavefunction analysis » Ehrenfest force » 2025-10-28 15:24:30
Dear Tian Lu, i'm currently in the process of verifying the correctness of this code. To do this, I needed to visualize this vector field, but I have into a problem: option 4 only allows to draw scalar fields and their gradients. Is there a way in multiwfn to directly visualize a vector field that is not the gradient of a scalar function? I couldn't find it in the manual
#3 Re: Multiwfn and wavefunction analysis » Ehrenfest force » 2025-10-27 18:07:43
real*8 function ehr(x,y,z)
use defvar
implicit real*8 (a-h,o-z)
real*8 wfnval(nmo),grad(3,nmo),hess(3,3,nmo),tens3(3,3,3,nmo)
real*8 F(3),phi,g(3),h(3,3),t3(3,3,3)
integer imo,i,j
real*8 occ,term,sumj
call orbderv(5,1,nmo,x,y,z,wfnval,grad,hess,tens3)
F=0D0
do imo=1,nmo
occ = MOocc(imo)
if (occ==0D0) cycle
phi = wfnval(imo)
g(:) = grad(:,imo)
h(:,:) = hess(:,:,imo)
t3(:,:,:) = tens3(:,:,:,imo)
do i=1,3
sumj = 0D0
do j=1,3
sumj = sumj + 2D0*(h(j,i)*g(j) + g(i)*h(j,j) - t3(j,i,j)*phi - h(i,j)*g(j))
end do
F(i) = F(i) - (-0.25D0*occ*sumj)
end do
end do
ehr = sqrt(F(1)**2 + F(2)**2 + F(3)**2)
end function
could you please check is it correct?
#4 Multiwfn and wavefunction analysis » Ehrenfest force » 2025-10-26 22:23:19
- Alexey
- Replies: 6
Hello, i'd like to study Ehrenfest force (both vector field and |F|). Multiwfn has a stress tensor, but is it possible to calculate the Ehrenfest force as its negative divergence? Does Multiwfn have such an option? I couldn't find it in the manual. (I understand that the Ehrenfest force derived from the stress tensor has problems due to the uncertainty of the stress tensor https://doi.org/10.1063/5.0174905 , but I'd still like to see it).
#5 Re: Multiwfn and wavefunction analysis » Topological analysis on grid without interpolation » 2025-10-13 19:18:29
Thanks, Tian Lu
#6 Multiwfn and wavefunction analysis » Topological analysis on grid without interpolation » 2025-10-12 20:10:32
- Alexey
- Replies: 2
Hello, I've discovered that the topological analysis based on a .cub file doesn't find all the bond paths (I'm using -3 BSpline interpolation). Therefore, I'd like to know if it's possible to perform a topological analysis based on a .cub file without interpolation, i.e., simply calculate the derivatives numerically on the grid?
I've attached an example where I couldn't find the bond paths between atoms 3 and 5 (water dimer) using
"High-quality grid, covering the entire system, about 1,728,000 points in total"
01_Water-Water_3_5.wfn
#7 Re: Multiwfn and wavefunction analysis » Topological analysis on grid » 2025-09-26 21:38:04
Dear Tian Lu, thank you for your reply. However, let me clarify my situation. I work with intermolecular interactions and want to search for critical points only between certain atoms. I'll give a simple example - the picture shows my blue box with a grid for atoms 4 and 15 (the Z axis is along atoms 4 and 15), it is 1 by 1 by 4 bohr in size, I changed the number of points in it from 32*32*32, 48*48*48 to 64*64*64, which seems to me to be a fairly dense grid. The thing is, with the standard generation settings, the bond path between atoms 4 and 15 appears only if I make 128*128*128 points in each direction of the box, which is too many. However, if I change the bond path generation settings to Stepsize, current: 0.2000, Stop generation if distance to any CP is smaller than: 0.2000, the bond path appears for a 64*64*64 grid and even 48*48*48 grids (see the other figure). My question is: is the bond path not displayed due to insufficient grid density or due to incorrect bond path generation settings for intermolecular interactions?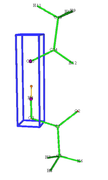
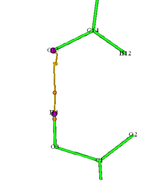
#8 Multiwfn and wavefunction analysis » Topological analysis on grid » 2025-09-25 17:34:34
- Alexey
- Replies: 3
Hello, I'm working with electron density on a fairly dense grid (more than 40,000 points at a size of 1 x 1 x 4 bohr box). When I try to perform a topological analysis, I can't generate a bond path between attractors properly. At first, I thought it was my grid, maybe it was too coarse, but it turned out to be related to the bond path generation settings. If I change the connection path generation settings, it might appear. I wanted to ask what settings are recommended for bond path generation and critical point detection when working with grid data. Thank you.
#9 Multiwfn and wavefunction analysis » Question about function which generates grid » 2025-09-19 11:31:07
- Alexey
- Replies: 1
Dear Tian Lu, thank you for your convenient program for working with wave functions. Multiwfn has a function
5
1
7
that constructs a grid of points centered between two specified atoms. As far as I understand, this box is always oriented along the coordinate axes in the original coordinate system specified in the input file. I was wondering if Multiwfn has a function that will not only center the box between two specified atoms, but also orient it along the AB vector (where A and B are the specified atomic indices). If there isn't such a function, could you please tell me which source code file I should change to achieve my goal? Thank you, Tian Lu
#10 Quantum Chemistry » T1 state optimization problem » 2025-08-30 13:54:25
- Alexey
- Replies: 1
Hello, I am trying to calculate the phosphorescence spectrum of a transition metal (copper) complex. I have attached the XYZ file. I am having a problem with DFT in Orca not being able to match the experimental phosphorescence result (2.83 eV). I have tried various functionals and found that using TDA improves the result for all the functionals I have tried, without it the triplet is greatly underestimated. However I am still not getting close to the experiment, my best result is 2.3 eV using wB97x/def2TZVP with TDA. LC-PBE and LC-BLYP give results around 2-2.2 eV, as do r2scan50, MN15, b3lyp and pbe0. Interestingly, using pbeh-3c I was getting 2.45 eV in the middle of an optimization run, but then the optimization broke down and I got a negative number. If anyone knows, please tell me which DFT functionals are recommended in my case, or should I still use multi-reference methods, thanks.
69
Cu 3.037023693 13.146441031 7.179215576
Cl 2.212517022 13.019565014 9.142739063
N 5.460505320 12.215472452 5.858057461
C 4.737813670 13.266919410 6.374465924
C 6.321793735 12.628308171 4.691855075
C 5.769806524 14.041468444 4.399304099
H 4.992384247 13.969811470 3.637328253
H 6.547421698 14.702735698 4.013817322
C 5.144976460 14.576079369 5.711217531
C 5.468031558 10.908300345 6.460193046
C 4.617158948 9.899965177 5.950828140
C 4.700314596 8.619200478 6.498765456
H 4.062138915 7.834819899 6.111859326
C 5.562131871 8.335214456 7.543670857
H 5.612756559 7.333139974 7.951822090
C 6.329109545 9.350242271 8.090153125
H 6.963209501 9.135023879 8.940720335
C 6.291429342 10.647734208 7.579745018
C 3.541889813 10.161505989 4.901721403
H 3.665117221 11.182473857 4.543599997
C 2.139646699 10.073868259 5.532174714
H 1.375245341 10.340955997 4.798348488
H 2.038426430 10.746566669 6.387285155
H 1.925676393 9.063385710 5.887593601
C 3.629097583 9.216767480 3.692682877
H 2.888402809 9.499055392 2.940581317
H 3.423514393 8.183054949 3.977798414
H 4.613035344 9.241438264 3.224101412
C 7.075666328 11.731049238 8.307280086
H 7.004023797 12.642358911 7.718998138
C 6.436914793 12.031905565 9.675286157
H 6.965220982 12.853351984 10.166149650
H 6.487981125 11.162557910 10.335085023
H 5.388588870 12.314051430 9.573834151
C 8.564625221 11.391161645 8.472085618
H 9.095447695 12.235174493 8.918856911
H 9.038791300 11.161479673 7.516681647
H 8.711107838 10.531929634 9.129756889
C 7.807353260 12.668661660 5.087242647
H 8.399786629 12.985542850 4.226109491
H 8.158375251 11.680282837 5.385368482
H 7.999922454 13.365150822 5.900903575
C 6.181231371 11.708480008 3.478098247
H 6.812851681 12.083522038 2.669929108
H 5.157336365 11.672620418 3.111120844
H 6.508416472 10.693754923 3.709117546
C 6.121169192 15.435427346 6.586107383
H 6.745723347 14.827638161 7.238726685
H 6.781368334 15.994282402 5.916081298
C 5.235637191 16.423881416 7.366265085
H 4.948816748 16.014410514 8.340261232
H 5.741514080 17.372868101 7.556254179
C 4.023288814 16.568846517 6.481537348
C 3.028266020 17.541177656 6.527953644
H 3.071761694 18.340464892 7.260169357
C 1.985403609 17.470556834 5.612908683
C 0.790972524 18.383643380 5.463097500
H 1.042834317 19.440923481 5.567261566
H 0.046854311 18.157352766 6.235616496
C 0.250683086 18.031995096 4.058342216
H 0.721769551 18.683107793 3.317818266
H -0.828096278 18.166260177 3.973064416
C 0.695845046 16.572642559 3.809607998
H -0.075423527 15.867616782 4.141658332
H 0.882216514 16.362581992 2.754305356
C 1.930513042 16.437031148 4.670824732
C 2.916640631 15.458776086 4.633254054
H 2.868087510 14.658422866 3.902751587
C 3.971711581 15.537324683 5.541719301
#11 Quantum Chemistry » ELF, LOL, laplacian » 2024-07-23 22:26:12
- Alexey
- Replies: 1
Hi everyone! Please recommend literature that reviews the topological analysis of the ELF, LOL and Laplacian functions. The meaning of the critical points (3;+1), (3;-1) and (3;+3) in the context of these functions is not entirely clear
#12 Multiwfn and wavefunction analysis » .wfn file » 2024-07-20 22:23:16
- Alexey
- Replies: 1
Will a function for outputting a .wfn file with promolecular wavefunction be added someday so that only xyz and "atomwfn" can be used to generate this file, and not a .wfn file with real wavefunction, calculated in the same basis as the atoms in "atomwfn"?
#13 Multiwfn and wavefunction analysis » promolecule » 2024-07-20 19:27:14
- Alexey
- Replies: 1
Hello! i need to use aspherical atomic density to build promolecular density, but i dont understand how to do it, because function 100(hidden option) > 10 give spherical avaraged density even if i use atom.wfn file with aspherical density. i rewrote subroutine sphatmraddens to get values for all (r;theta;phi) points, but i dont understand how to use this data for building promolecular density. i attach my code and file density.txt for some atom which i get with the help of my code. should i create new lagintpol in (r,theta,phi) space to use my data in building promolecular density with calcprodens(x,y,z,0)? or what? help me please
subroutine sphatmraddens
use defvar
use util
use functions
implicit real*8 (a-h,o-z)
! Declare variables
real*8, allocatable :: radx(:), rady(:), radz(:), radpos(:), density(:)
integer :: ntheta, nphi
real*8 :: theta, phi
integer :: irad, itheta, iphi, idx
real*8 :: rad, x, y, z
integer :: ifinish, iprogstp, iprogcrit, nradpt
integer :: itmp
parameter (truncrho = 1D-8, rlow = 0D0, rhigh = 12D0, nradpt = 200, ntheta = 50, nphi = 100)
! Allocate arrays
allocate(radx(nradpt), rady(nradpt), radz(nradpt), radpos(nradpt), density(nradpt*ntheta*nphi))
ifinish = 0
iprogstp = 20
iprogcrit = iprogstp
write(*,*) "Calculating..."
! Parallel loop
!$OMP PARALLEL DO SHARED(density, radpos, ifinish, iprogcrit) PRIVATE(irad, itheta, iphi, rad, x, y, z, theta, phi, idx) schedule(dynamic) NUM_THREADS(nthreads)
do irad = 1, nradpt
rad = rlow + (rhigh - rlow) * (irad - 1) / (nradpt - 1)
radpos(irad) = rad
do itheta = 0, ntheta-1
theta = pi * itheta / (ntheta - 1) ! theta ranges from 0 to pi
do iphi = 0, nphi-1
phi = 2 * pi * iphi / nphi ! phi ranges from 0 to 2*pi
x = rad * sin(theta) * cos(phi)
y = rad * sin(theta) * sin(phi)
z = rad * cos(theta)
idx = (irad - 1) * ntheta * nphi + itheta * nphi + iphi + 1
! Compute density
density(idx) = fdens(x, y, z)
end do
end do
ifinish = ifinish + 1
if (ifinish >= iprogcrit) then
call showprog(ifinish, nradpt)
iprogcrit = iprogcrit + iprogstp
end if
end do
!$OMP END PARALLEL DO
! Output results
open(10, file="density.txt", status="replace")
itmp = 0
do irad = 1, nradpt
do itheta = 0, ntheta-1
theta = pi * itheta / (ntheta - 1)
do iphi = 0, nphi-1
phi = 2 * pi * iphi / nphi
idx = (irad - 1) * ntheta * nphi + itheta * nphi + iphi + 1
if (density(idx) > truncrho) then
itmp = itmp + 1
write(10, "(' r=', f12.6, ' theta=', f12.6, ' phi=', f12.6, ' density=', f25.10, 'D0')") radpos(irad), theta, phi, density(idx)
end if
end do
end do
end do
close(10)
write(*,*) "The result has been output to density.txt in the current folder"
end subroutine sphatmraddensi get correct values for all (r,theta,phi) points with this code, but i need to construct density with this values (do not pay attention that the density values are small, in my case it should be so)
file: https://drive.google.com/file/d/136n0jN … sp=sharing
p.s. may be my way to build promolecular density with aspherical atomic densities is bad? is there other way to do it?
i need to use molecule.xyz input and then analyze promolecular density, so 1000>17 doesnt suit me because i need molecule.wfn file for it
#14 Multiwfn and wavefunction analysis » modify function » 2024-07-08 16:30:00
- Alexey
- Replies: 1
i need to modify this part of 'calchessmat_prodens' subroutine to get good promolecular density and calculate its derivatives for all elements. the following part of code doesnt calculate derivatives in the nuclear positions (but value of promolecular density is good calculated), however, in non-nuclear positions the derivative is calculated normally, what should I write in the following code to get derivatives in nuclear positionsn? (i ask this because if i use "output prop in point" for the iuserfunc==-2 function then i get derivatives at nuclear positions, but in the case below (elerho from my new calchessmat_prodens) i don't get derivatives of elerho in nuclear positions, only value)
if (iele>=1) then
if (r>atmrhocut(iele)) cycle
call genatmraddens(iele,rhoarr,npt) !Extract spherically averaged radial density of corresponding element at specific grids
if (idohess==0) then
call lagintpol(atmradpos(1:npt),rhoarr(1:npt),npt,r,term,der1r,der2r,2)
else if (idohess==1) then
call lagintpol(atmradpos(1:npt),rhoarr(1:npt),npt,r,term,der1r,der2r,3)
end if
elerho=elerho+term
der1rdr=der1r/r
derx=derx+der1rdr*rx
dery=dery+der1rdr*ry
derz=derz+der1rdr*rz
if (idohess==1) then
tmpval=(der2r-der1rdr)/r2
dxx=dxx+der1rdr+tmpval*rx2
dyy=dyy+der1rdr+tmpval*ry2
dzz=dzz+der1rdr+tmpval*rz2
dxy=dxy+tmpval*rx*ry
dyz=dyz+tmpval*ry*rz
dxz=dxz+tmpval*rx*rz
end if
end if#15 Re: Multiwfn and wavefunction analysis » function.f90 » 2024-07-07 16:16:58
thank you soooo much
but so, im following to your advice to modife calchessmat_prodens code to generate good promoldens
i did this:
(i changed 'if ele<=18' to 'if ele >=118)
subroutine calchessmat_prodens(xin,yin,zin,elerho,elegrad,elehess)
use util
real*8 elerho,xin,yin,zin
real*8,optional :: elegrad(3),elehess(3,3)
real*8 posarr(200),rhoarr(200),tvec(3)
elerho=0D0
derx=0D0
dery=0D0
derz=0D0
dxx=0D0
dyy=0D0
dzz=0D0
dxy=0D0
dyz=0D0
dxz=0D0
idohess=0
if (present(elehess)) idohess=1
call getpointcell(xin,yin,zin,ic,jc,kc)
do icell=ic-PBCnx,ic+PBCnx
do jcell=jc-PBCny,jc+PBCny
do kcell=kc-PBCnz,kc+PBCnz
call tvec_PBC(icell,jcell,kcell,tvec)
do i=1,nfragatm
iatm=fragatm(i)
iele=a(iatm)%index
!rx=a(iatm)%x+tvec(1)-xin !Wrong code, older than 2022-Sep-18
!ry=a(iatm)%y+tvec(2)-yin
!rz=a(iatm)%z+tvec(3)-zin
rx=xin-tvec(1)-a(iatm)%x !Relative x
ry=yin-tvec(2)-a(iatm)%y
rz=zin-tvec(3)-a(iatm)%z
rx2=rx*rx
ry2=ry*ry
rz2=rz*rz
r2=rx2+ry2+rz2
r=dsqrt(r2)
if (iele>=118) then !H~Ar, use Weitao Yang's fitted parameters as original RDG paper
if (atomdenscut==1) then !Tight cutoff, for CHNO corresponding to cutoff at rho=0.00001
if (iele==1.and.r2>25D0) then !H, 6.63^2=43.9569. But this seems to be unnecessarily large, so I use 5^2=25
cycle
else if (iele==6.and.r2>58.6756D0) then !C, 7.66^2=58.6756
cycle
else if (iele==7.and.r2>43.917129D0) then !N, 6.627^2=43.917129
cycle
else if (iele==8.and.r2>34.9281D0) then !O, 5.91^2=34.9281
cycle
else if (r2>(2.5D0*vdwr(iele))**2) then !Other cases, larger than 2.5 times of its vdw radius will be skipped
cycle
end if
else if (atomdenscut==2) then !Medium cutoff, the result may be not as accurate as atomdenscut==1, but much more cheaper
if (r2>(2.2D0*vdwr(iele))**2) cycle
else if (atomdenscut==3) then !Loose cutoff, the most inaccurate
if (r2>(1.8D0*vdwr(iele))**2) cycle
else if (atomdenscut==4) then !Foolish cutoff, you need to know what you are doing
if (r2>(1.5D0*vdwr(iele))**2) cycle
end if
r2_1d5=r2**1.5D0
do iSTO=1,3
if (YWTatomcoeff(iele,iSTO)==0D0) cycle
expterm=YWTatomexp(iele,iSTO)
term=YWTatomcoeff(iele,iSTO)*dexp(-r/expterm)
elerho=elerho+term
if (r==0D0) cycle !Derivative of STO at nuclei is pointless
tmp=term/expterm/r
derx=derx-tmp*rx !Calculating gradient doesn't cost detectable time, so always calculate it
dery=dery-tmp*ry
derz=derz-tmp*rz
if (idohess==1) then
tmp1=1/r2_1d5/expterm
tmp2=1/r2/(expterm*expterm)
dxx=dxx+term*(tmp1*rx2-1/r/expterm+tmp2*rx2)
dyy=dyy+term*(tmp1*ry2-1/r/expterm+tmp2*ry2)
dzz=dzz+term*(tmp1*rz2-1/r/expterm+tmp2*rz2)
tmp=term*(tmp1+tmp2)
dxy=dxy+rx*ry*tmp
dyz=dyz+ry*rz*tmp
dxz=dxz+rx*rz*tmp
end if
end do
else !Heavier than Ar
if (r>atmrhocut(iele)) cycle
call genatmraddens(iele,rhoarr,npt) !Extract spherically averaged radial density of corresponding element at specific grids
if (idohess==0) then
call lagintpol(atmradpos(1:npt),rhoarr(1:npt),npt,r,term,der1r,der2r,2)
else if (idohess==1) then
call lagintpol(atmradpos(1:npt),rhoarr(1:npt),npt,r,term,der1r,der2r,3)
end if
elerho=elerho+term
der1rdr=der1r/r
derx=derx+der1rdr*rx
dery=dery+der1rdr*ry
derz=derz+der1rdr*rz
if (idohess==1) then !See promolecular_grid routine in props.f90 of NCIplot
tmpval=(der2r-der1rdr)/r2
dxx=dxx+der1rdr+tmpval*rx2
dyy=dyy+der1rdr+tmpval*ry2
dzz=dzz+der1rdr+tmpval*rz2
dxy=dxy+tmpval*rx*ry
dyz=dyz+tmpval*ry*rz
dxz=dxz+tmpval*rx*rz
end if
end if
end do
end do
end do
end do
if (present(elegrad)) then
elegrad(1)=derx
elegrad(2)=dery
elegrad(3)=derz
end if
if (idohess==1) then
elehess(1,1)=dxx
elehess(2,2)=dyy
elehess(3,3)=dzz
elehess(1,2)=dxy
elehess(2,3)=dyz
elehess(1,3)=dxz
elehess(2,1)=dxy
elehess(3,2)=dyz
elehess(3,1)=dxz
end if
end subroutineand if i try to "Output all properties at a point" (point is the O atom in H2O) i get follow for promolecular density (promolecular density is good calculated, but its derivatives are not calculated)
Density of electrons: 0.3441456709E+00
Reduced density gradient: 0.1000000000E+03
Note: Matrix diagonalization exceed max cycle before convergence
Sign(lambda2)*rho: NaN
ESP from nuclear charges: 0.1000000000E+04
van der Waals potential (probe atom: C ): 0.1280973043+126 kcal/mol
User-defined real space function: NaN
Note: Below information are for electron density
Components of gradient in x/y/z are:
NaN NaN NaN
Norm of gradient is: NaN
Components of Laplacian in x/y/z are:
NaN NaN NaN
Total: NaN
Hessian matrix:
NaN NaN NaN
NaN NaN NaN
NaN NaN NaN#16 Re: Multiwfn and wavefunction analysis » function.f90 » 2024-07-07 14:35:49
I create my own function that creates promolecular density (input file xyz) with calcprodens(x,y,z,0) and then analyze it. should i use subroutine gencalchessmat to calc promolgrad and hess?
#17 Re: Multiwfn and wavefunction analysis » function.f90 » 2024-07-07 11:01:37
sorry))
is it possible to use calcprodens(x,y,z,0) to generate "good" promolecular density and then analyze it? And could you please tell me, what function/subroutine is used for topological analysis of iuserfunc==-2(calcprodens) How are derivatives and the Hessian matrix calculated?
#18 Re: Multiwfn and wavefunction analysis » function.f90 » 2024-07-07 10:20:04
а нельзя использовать функцию calcprodens(x,y,z,0) для генерации хорошей промолекулярной плотности а потом использовать ее для анализа? And could you please tell me, щn the basis of what function is topological analysis done iuserfunc==-2(calcprodens) How are derivatives and the Hessian matrix calculated?
#19 Multiwfn and wavefunction analysis » function.f90 » 2024-07-06 13:19:18
- Alexey
- Replies: 9
could you please explain to me what is the difference between calchessmat_dens_promol and calchessmat_prodens? which function i need to use to calculate promolecular density with built-in spherical atomic densities and then analyze it (calculate gradient, lapl on promolecular density)?
Pages: 1